- Record: found
- Abstract: found
- Article: found
Molecular Analysis, Pathogenic Mechanisms, and Readthrough Therapy on a Large Cohort of Kabuki Syndrome Patients
Read this article at
Abstract
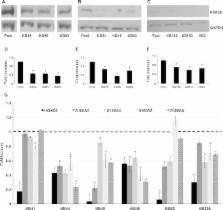
Kabuki syndrome (KS) is a multiple congenital anomalies syndrome characterized by characteristic facial features and varying degrees of mental retardation, caused by mutations in KMT2D/ MLL2 and KDM6A/ UTX genes. In this study, we performed a mutational screening on 303 Kabuki patients by direct sequencing, MLPA, and quantitative PCR identifying 133 KMT2D, 62 never described before, and four KDM6A mutations, three of them are novel. We found that a number of KMT2D truncating mutations result in mRNA degradation through the nonsense-mediated mRNA decay, contributing to protein haploinsufficiency. Furthermore, we demonstrated that the reduction of KMT2D protein level in patients’ lymphoblastoid and skin fibroblast cell lines carrying KMT2D-truncating mutations affects the expression levels of known KMT2D target genes. Finally, we hypothesized that the KS patients may benefit from a readthrough therapy to restore physiological levels of KMT2D and KDM6A proteins. To assess this, we performed a proof-of-principle study on 14 KMT2D and two KDM6A nonsense mutations using specific compounds that mediate translational readthrough and thereby stimulate the re-expression of full-length functional proteins. Our experimental data showed that both KMT2D and KDM6A nonsense mutations displayed high levels of readthrough in response to gentamicin treatment, paving the way to further studies aimed at eventually treating some Kabuki patients with readthrough inducers.
Related collections
Most cited references39
- Record: found
- Abstract: found
- Article: not found
Improved splice site detection in Genie.
- Record: found
- Abstract: found
- Article: not found
Estrogen receptors and human disease.
- Record: found
- Abstract: found
- Article: not found