- Record: found
- Abstract: found
- Article: found
Overexpression of MYB drives proliferation of CYLD‐defective cylindroma cells†
Read this article at
Abstract
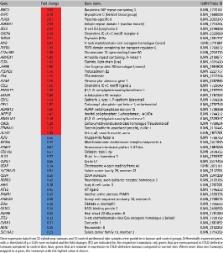
Cutaneous cylindroma is an adnexal tumour with apocrine differentiation. A predisposition to multiple cylindromas is seen in patients with Brooke–Spiegler syndrome, who carry germline mutations in the tumour suppressor gene CYLD . Previous studies of inherited cylindromas have highlighted the frequent presence of bi‐allelic truncating CYLD mutations as a recurrent driver mutation. We have previously shown that sporadic cylindromas express either MYB–NFIB fusion transcripts or show evidence of MYB activation in the absence of such fusions. Here, we investigated inherited cylindromas from several families with germline CYLD mutations for the presence of MYB activation. Strikingly, none of the inherited CYLD ‐defective ( n = 23) tumours expressed MYB–NFIB fusion transcripts. However, MYB expression was increased in the majority of tumours (69%) and global gene expression analysis revealed that well‐established MYB target genes were up‐regulated in CYLD ‐defective tumours. Moreover, knock‐down of MYB expression caused a significant reduction in cylindroma cell proliferation, suggesting that MYB is also a key player and oncogenic driver in inherited cylindromas. Taken together, our findings suggest molecular heterogeneity in the pathogenesis of sporadic and inherited cutaneous cylindromas, with convergence on MYB activation. © 2016 The Authors. The Journal of Pathology published by John Wiley & Sons Ltd on behalf of Pathological Society of Great Britain and Ireland.
Related collections
Most cited references16
- Record: found
- Abstract: found
- Article: not found
Cyld inhibits tumor cell proliferation by blocking Bcl-3-dependent NF-kappaB signaling.
- Record: found
- Abstract: found
- Article: not found
Whole exome sequencing of adenoid cystic carcinoma.
- Record: found
- Abstract: found
- Article: not found