- Record: found
- Abstract: found
- Article: not found
Fatty acid transporter 2 reprograms neutrophils in cancer

Summary
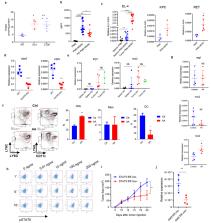
Polymorphonuclear myeloid derived suppressor cells (PMN-MDSC) are pathologically activated neutrophils that are critically important for the regulation of immune responses in cancer. They contribute to the failure of cancer therapies and are associated with poor clinical outcomes. Despite the recent advances in understanding of the PMN-MDSC biology, the mechanisms responsible for pathological activation of neutrophils are not well defined, which limits selective targeting of these cells. Here, we report that mouse and human PMN-MDSC exclusively up-regulate fatty acid transporter protein 2 (FATP2). Over-expression of FATP2 in PMN-MDSC was controlled by GM-CSF, through the activation of STAT5 transcription factor. Deletion of FATP2 abrogated the suppressive activity of PMN-MDSC. The main mechanism of FATP2 mediated suppressive activity involved uptake of arachidonic acid (AA) and synthesis of prostaglandin E2 (PGE2). The selective pharmacological inhibition of FATP2 abrogated the activity of PMN-MDSC and substantially delayed tumor progression. In combination with check-point inhibitors it blocked tumor progression in mice. Thus, FATP2 mediates acquisition of immune suppressive activity by PMN-MDSC and represents a new target to selectively inhibit the functions of PMN-MDSC and improve the effect of cancer therapy.
Related collections
Most cited references19
- Record: found
- Abstract: found
- Article: not found
ER Stress Sensor XBP1 Controls Anti-tumor Immunity by Disrupting Dendritic Cell Homeostasis.
- Record: found
- Abstract: found
- Article: not found
Lipid accumulation and dendritic cell dysfunction in cancer
- Record: found
- Abstract: found
- Article: not found