- Record: found
- Abstract: found
- Article: not found
PINK1 Is Necessary for Long Term Survival and Mitochondrial Function in Human Dopaminergic Neurons
Read this article at

Abstract
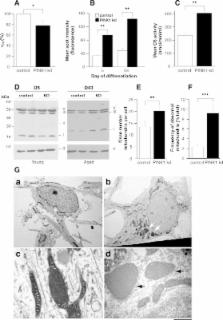
Parkinson's disease (PD) is a common age-related neurodegenerative disease and it is critical to develop models which recapitulate the pathogenic process including the effect of the ageing process. Although the pathogenesis of sporadic PD is unknown, the identification of the mendelian genetic factor PINK1 has provided new mechanistic insights. In order to investigate the role of PINK1 in Parkinson's disease, we studied PINK1 loss of function in human and primary mouse neurons. Using RNAi, we created stable PINK1 knockdown in human dopaminergic neurons differentiated from foetal ventral mesencephalon stem cells, as well as in an immortalised human neuroblastoma cell line. We sought to validate our findings in primary neurons derived from a transgenic PINK1 knockout mouse. For the first time we demonstrate an age dependent neurodegenerative phenotype in human and mouse neurons. PINK1 deficiency leads to reduced long-term viability in human neurons, which die via the mitochondrial apoptosis pathway. Human neurons lacking PINK1 demonstrate features of marked oxidative stress with widespread mitochondrial dysfunction and abnormal mitochondrial morphology. We report that PINK1 plays a neuroprotective role in the mitochondria of mammalian neurons, especially against stress such as staurosporine. In addition we provide evidence that cellular compensatory mechanisms such as mitochondrial biogenesis and upregulation of lysosomal degradation pathways occur in PINK1 deficiency. The phenotypic effects of PINK1 loss-of-function described here in mammalian neurons provides mechanistic insight into the age-related degeneration of nigral dopaminergic neurons seen in PD.
Related collections
Most cited references49
- Record: found
- Abstract: found
- Article: not found
ROS, mitochondria and the regulation of autophagy.
- Record: found
- Abstract: found
- Article: not found
The PINK1/Parkin pathway regulates mitochondrial morphology.
- Record: found
- Abstract: found
- Article: not found