- Record: found
- Abstract: found
- Article: found
Is the Infection of the SARS-CoV-2 Delta Variant Associated With the Outcomes of COVID-19 Patients?
Read this article at
Abstract
Background: Severe acute respiratory syndrome Coronavirus 2 (SARS-CoV-2) Delta variant (B.1.617.2) has been responsible for the current increase in Coronavirus disease 2019 (COVID-19) infectivity rate worldwide. We compared the impact of the Delta variant and non-Delta variant on the COVID-19 outcomes in patients from Yogyakarta and Central Java provinces, Indonesia.
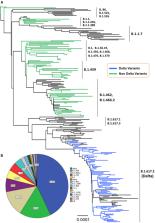
Methods: In this cross-sectional study, we ascertained 161 patients, 69 with the Delta variant and 92 with the non-Delta variant. The Illumina MiSeq next-generation sequencer was used to perform the whole-genome sequences of SARS-CoV-2.
Results: The mean age of patients with the Delta variant and the non-Delta variant was 27.3 ± 20.0 and 43.0 ± 20.9 ( p = 3 × 10 −6). The patients with Delta variant consisted of 23 males and 46 females, while the patients with the non-Delta variant involved 56 males and 36 females ( p = 0.001). The Ct value of the Delta variant (18.4 ± 2.9) was significantly lower than that of the non-Delta variant (19.5 ± 3.8) ( p = 0.043). There was no significant difference in the hospitalization and mortality of patients with Delta and non-Delta variants ( p = 0.80 and 0.29, respectively). None of the prognostic factors were associated with the hospitalization, except diabetes with an OR of 3.6 (95% CI = 1.02–12.5; p = 0.036). Moreover, the patients with the following factors have been associated with higher mortality rate than the patients without the factors: age ≥65 years, obesity, diabetes, hypertension, and cardiovascular disease with the OR of 11 (95% CI = 3.4–36; p = 8 × 10 −5), 27 (95% CI = 6.1–118; p = 1 × 10 −5), 15.6 (95% CI = 5.3–46; p = 6 × 10 −7), 12 (95% CI = 4–35.3; p = 1.2 × 10 −5), and 6.8 (95% CI = 2.1–22.1; p = 0.003), respectively. Multivariate analysis showed that age ≥65 years, obesity, diabetes, and hypertension were the strong prognostic factors for the mortality of COVID-19 patients with the OR of 3.6 (95% CI = 0.58–21.9; p = 0.028), 16.6 (95% CI = 2.5–107.1; p = 0.003), 5.5 (95% CI = 1.3–23.7; p = 0.021), and 5.8 (95% CI = 1.02–32.8; p = 0.047), respectively.
Conclusions: We show that the patients infected by the SARS-CoV-2 Delta variant have a lower Ct value than the patients infected by the non-Delta variant, implying that the Delta variant has a higher viral load, which might cause a more transmissible virus among humans. However, the Delta variant does not affect the COVID-19 outcomes in our patients. Our study also confirms that older age and comorbidity increase the mortality rate of patients with COVID-19.
Related collections
Most cited references37
- Record: found
- Abstract: found
- Article: not found
MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms.
- Record: found
- Abstract: found
- Article: not found
A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences.
- Record: found
- Abstract: found
- Article: not found