- Record: found
- Abstract: found
- Article: found
Novel heterozygous mutations of SLC12A3 gene in a Chinese pedigree with Gitelman syndrome: A care-compliant case report

Read this article at
Abstract
Rationale:
The diagnosis of Gentleman syndrome (GS) is usually delayed because the clinical symptoms are easily mistaken.
Patient concerns:
A 19-year-old male patient was referred to endocrinology due to intermittent twitch of extremities for approximately 7 years.
Diagnoses:
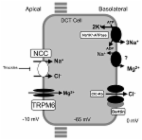
The diagnosis of GS was made based on the laboratory and gene detection results. We identified 2 new variants in the SLC12A3 gene [c.857 A > C (exon7) and c.2089_2095del (exon17)] in his Asian family.
Interventions:
The patient received the treatment of potassium chloride sustained release tablets, potassium magnesium aspartate and spironolactone. After given potassium supplement through enema, his serum potassium level was corrected to normal.
Outcomes:
The electrolyte imbalance including hypokalemia and hypomagnesemia were improved with a remission of the clinical manifestations. But the patient’s condition still could not remain stable for his irregular oral potassium supplementation during the follow-up of nearly 3 months.
Lessons:
Our finding broadens the variant spectrum of SLC12A3 and contributes to a more quickly genetic counseling. As a result, when a patient presents with persistent, unspecified, and inadequately treated hypokalemia, tests for GS should indeed be considered. For suspected cases of GS, genetic testing should always be considered in the diagnosis.
Related collections
Most cited references12
- Record: found
- Abstract: found
- Article: found
Gitelman syndrome: consensus and guidance from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference
- Record: found
- Abstract: found
- Article: found