- Record: found
- Abstract: found
- Article: found
Rapid and accurate alignment of nucleotide conversion sequencing reads with HISAT-3N
Read this article at
Abstract
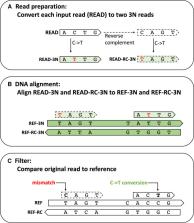
Sequencing technologies using nucleotide conversion techniques such as cytosine to thymine in bisulfite-seq and thymine to cytosine in SLAM seq are powerful tools to explore the chemical intricacies of cellular processes. To date, no one has developed a unified methodology for aligning converted sequences and consolidating alignment of these technologies in one package. In this paper, we describe hierarchical indexing for spliced alignment of transcripts–3 nucleotides (HISAT-3N), which can rapidly and accurately align sequences consisting of any nucleotide conversion by leveraging the powerful hierarchical index and repeat index algorithms originally developed for the HISAT software. Tests on real and simulated data sets show that HISAT-3N is faster than other modern systems, with greater alignment accuracy, higher scalability, and smaller memory requirements. HISAT-3N therefore becomes an ideal aligner when used with converted sequence technologies.
Related collections
Most cited references26

- Record: found
- Abstract: found
- Article: found
The Sequence Alignment/Map format and SAMtools
- Record: found
- Abstract: found
- Article: not found
STAR: ultrafast universal RNA-seq aligner.
- Record: found
- Abstract: found
- Article: not found