- Record: found
- Abstract: found
- Article: found
OptForce: An Optimization Procedure for Identifying All Genetic Manipulations Leading to Targeted Overproductions
Read this article at
Abstract
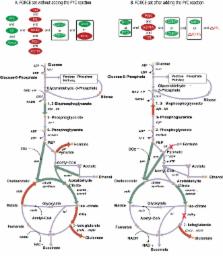
Computational procedures for predicting metabolic interventions leading to the overproduction of biochemicals in microbial strains are widely in use. However, these methods rely on surrogate biological objectives (e.g., maximize growth rate or minimize metabolic adjustments) and do not make use of flux measurements often available for the wild-type strain. In this work, we introduce the OptForce procedure that identifies all possible engineering interventions by classifying reactions in the metabolic model depending upon whether their flux values must increase, decrease or become equal to zero to meet a pre-specified overproduction target. We hierarchically apply this classification rule for pairs, triples, quadruples, etc. of reactions. This leads to the identification of a sufficient and non-redundant set of fluxes that must change (i.e., MUST set) to meet a pre-specified overproduction target. Starting with this set we subsequently extract a minimal set of fluxes that must actively be forced through genetic manipulations (i.e., FORCE set) to ensure that all fluxes in the network are consistent with the overproduction objective. We demonstrate our OptForce framework for succinate production in Escherichia coli using the most recent in silico E. coli model, iAF1260. The method not only recapitulates existing engineering strategies but also reveals non-intuitive ones that boost succinate production by performing coordinated changes on pathways distant from the last steps of succinate synthesis.
Author Summary
Over the past few years, there has been an unprecedented increase in the use of microorganisms for the production of biofuels, industrial chemicals and pharmaceutical precursors. In this regard, biotechnologists are confronted with the challenge to efficiently convert biomass and other renewable resources into useful biochemicals. With the advent of organism-specific mathematical models of metabolism, scientists have used computations to identify genetic modifications that maximize the yield of a desired product. In this paper, we introduce OptForce, an algorithm that identifies all possible metabolic interventions that lead to the overproduction of a biochemical of interest. Unlike existing techniques, OptForce does not rely on the maximization of a fitness function to predict metabolic fluxes. Instead, OptForce contrasts the metabolic flux patterns observed in an initial strain and a strain overproducing the chemical at the target yield. The essence of this procedure is the identification of all coordinated reaction modifications that force the network towards the overproduction target. We used OptForce to predict metabolic interventions for succinate overproduction in Escherichia coli. The results described in this paper not only uncover existing strain designs for succinate production but also elucidate new ones that can be experimentally explored.
Related collections
Most cited references54

- Record: found
- Abstract: found
- Article: found
A genome-scale metabolic reconstruction for Escherichia coli K-12 MG1655 that accounts for 1260 ORFs and thermodynamic information
- Record: found
- Abstract: found
- Article: not found
The effects of alternate optimal solutions in constraint-based genome-scale metabolic models.
- Record: found
- Abstract: found
- Article: not found