
- Record: found
- Abstract: found
- Article: found
Deterministic Evolutionary Trajectories Influence Primary Tumor Growth: TRACERx Renal

Summary
The evolutionary features of clear-cell renal cell carcinoma (ccRCC) have not been systematically studied to date. We analyzed 1,206 primary tumor regions from 101 patients recruited into the multi-center prospective study, TRACERx Renal. We observe up to 30 driver events per tumor and show that subclonal diversification is associated with known prognostic parameters. By resolving the patterns of driver event ordering, co-occurrence, and mutual exclusivity at clone level, we show the deterministic nature of clonal evolution. ccRCC can be grouped into seven evolutionary subtypes, ranging from tumors characterized by early fixation of multiple mutational and copy number drivers and rapid metastases to highly branched tumors with >10 subclonal drivers and extensive parallel evolution associated with attenuated progression. We identify genetic diversity and chromosomal complexity as determinants of patient outcome. Our insights reconcile the variable clinical behavior of ccRCC and suggest evolutionary potential as a biomarker for both intervention and surveillance.
Graphical Abstract
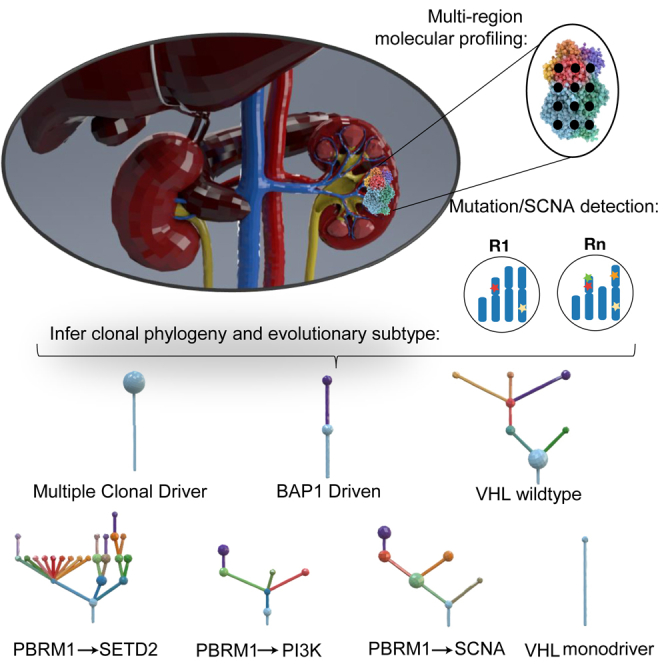
Highlights
-
•
ccRCC evolutionary subtypes correlate with clinical phenotypes
-
•
Genetic diversity and chromosome complexity contribute to patient outcomes
-
•
Early fixation of multiple driver events leads to rapid growth and metastases
-
•
Subclonal diversification is linked with slower growth and attenuated metastases
Abstract
A multi-center prospective study on 101 patients with clear-cell renal cell carcinoma resolves the evolutionary features and subtypes underpinning the diverse clinical phenotypes of the disease and suggests these features as potential biomarkers for guiding intervention and surveillance.
Related collections
Most cited references69

- Record: found
- Abstract: found
- Article: found
Fast and accurate short read alignment with Burrows–Wheeler transform

- Record: found
- Abstract: found
- Article: found
BEDTools: a flexible suite of utilities for comparing genomic features

- Record: found
- Abstract: found
- Article: found