
- Record: found
- Abstract: found
- Article: found
NMR and DFT investigations of structure of colchicine in various solvents including density functional theory calculations
Read this article at
Abstract
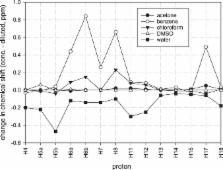
A detailed NMR investigation of the chemical shifts of hydrogen and carbon atoms associated with the structure of the naturally occurring alkaloid colchicine was conducted using high field NMR. Initially, the experimental chemical shifts for colchicine in chloroform and DMSO were compared to the values calculated using density functional theory (DFT). There were significant deviations observed for the chloroform solvent, but these were only slight in the DMSO solution. Dilution of the chloroform solution changed the experimental chemical shifts and improved agreement with the DFT calculations, suggesting self-aggregation at higher concentrations. A dimeric model was proposed for which agreement with the DFT calculated chemical shifts was better than for corresponding monomeric structures. Three further solvents were studied to evaluate changes in chemical shift values at different dilutions. Chloroform, benzene and water showed significant chemical shift changes implying self-aggregation, whereas DMSO and acetone did not show significant change upon dilution.
Related collections
Most cited references22
- Record: found
- Abstract: not found
- Article: not found
The IEF version of the PCM solvation method: an overview of a new method addressed to study molecular solutes at the QM ab initio level
- Record: found
- Abstract: not found
- Article: not found
Computational prediction of 1H and 13C chemical shifts: a useful tool for natural product, mechanistic, and synthetic organic chemistry.
- Record: found
- Abstract: found
- Article: not found