- Record: found
- Abstract: found
- Article: found
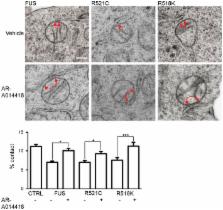
ALS/FTD‐associated FUS activates GSK‐3β to disrupt the VAPB–PTPIP51 interaction and ER–mitochondria associations
Read this article at
Abstract
Defective FUS metabolism is strongly associated with amyotrophic lateral sclerosis and frontotemporal dementia ( ALS/ FTD), but the mechanisms linking FUS to disease are not properly understood. However, many of the functions disrupted in ALS/ FTD are regulated by signalling between the endoplasmic reticulum ( ER) and mitochondria. This signalling is facilitated by close physical associations between the two organelles that are mediated by binding of the integral ER protein VAPB to the outer mitochondrial membrane protein PTPIP51, which act as molecular scaffolds to tether the two organelles. Here, we show that FUS disrupts the VAPB– PTPIP51 interaction and ER–mitochondria associations. These disruptions are accompanied by perturbation of Ca 2+ uptake by mitochondria following its release from ER stores, which is a physiological read‐out of ER–mitochondria contacts. We also demonstrate that mitochondrial ATP production is impaired in FUS‐expressing cells; mitochondrial ATP production is linked to Ca 2+ levels. Finally, we demonstrate that the FUS‐induced reductions to ER–mitochondria associations and are linked to activation of glycogen synthase kinase‐3β ( GSK‐3β), a kinase already strongly associated with ALS/ FTD.
Related collections
Most cited references22
- Record: found
- Abstract: found
- Article: not found
A mutation in sigma-1 receptor causes juvenile amyotrophic lateral sclerosis.
- Record: found
- Abstract: found
- Article: found
ALS mutant FUS disrupts nuclear localization and sequesters wild-type FUS within cytoplasmic stress granules
- Record: found
- Abstract: found
- Article: not found