- Record: found
- Abstract: found
- Article: found
Characterization of the Uncertainty of Divergence Time Estimation under Relaxed Molecular Clock Models Using Multiple Loci
Read this article at
Abstract
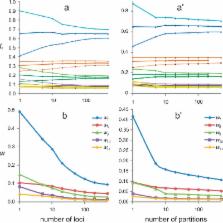
Genetic sequence data provide information about the distances between species or branch lengths in a phylogeny, but not about the absolute divergence times or the evolutionary rates directly. Bayesian methods for dating species divergences estimate times and rates by assigning priors on them. In particular, the prior on times (node ages on the phylogeny) incorporates information in the fossil record to calibrate the molecular tree. Because times and rates are confounded, our posterior time estimates will not approach point values even if an infinite amount of sequence data are used in the analysis. In a previous study we developed a finite-sites theory to characterize the uncertainty in Bayesian divergence time estimation in analysis of large but finite sequence data sets under a strict molecular clock. As most modern clock dating analyses use more than one locus and are conducted under relaxed clock models, here we extend the theory to the case of relaxed clock analysis of data from multiple loci (site partitions). Uncertainty in posterior time estimates is partitioned into three sources: Sampling errors in the estimates of branch lengths in the tree for each locus due to limited sequence length, variation of substitution rates among lineages and among loci, and uncertainty in fossil calibrations. Using a simple but analogous estimation problem involving the multivariate normal distribution, we predict that as the number of loci ( ) goes to infinity, the variance in posterior time estimates decreases and approaches the infinite-data limit at the rate of 1/ , and the limit is independent of the number of sites in the sequence alignment. We then confirmed the predictions by using computer simulation on phylogenies of two or three species, and by analyzing a real genomic data set for six primate species. Our results suggest that with the fossil calibrations fixed, analyzing multiple loci or site partitions is the most effective way for improving the precision of posterior time estimation. However, even if a huge amount of sequence data is analyzed, considerable uncertainty will persist in time estimates.
Related collections
Most cited references26
- Record: found
- Abstract: found
- Article: not found
Dating of the human-ape splitting by a molecular clock of mitochondrial DNA.

- Record: found
- Abstract: found
- Article: found
A Total-Evidence Approach to Dating with Fossils, Applied to the Early Radiation of the Hymenoptera
- Record: found
- Abstract: found
- Article: not found