- Record: found
- Abstract: found
- Article: found
Hepatic protein-tyrosine phosphatase 1B disruption and pharmacological inhibition attenuate ethanol-induced oxidative stress and ameliorate alcoholic liver disease in mice

Read this article at
Abstract
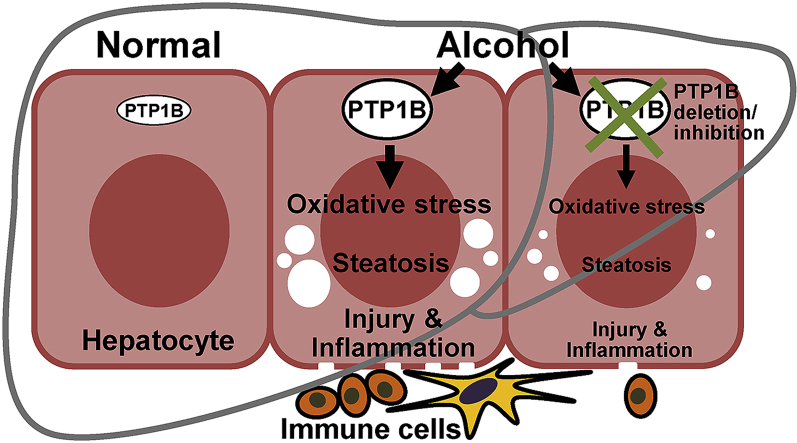
Alcoholic liver disease (ALD) is a major health problem and a significant cause of liver-related death. Currently, the mainstay for ALD therapy is alcohol abstinence highlighting the need to develop pharmacotherapeutic approaches. Protein-tyrosine phosphatase 1B (PTP1B) is an established regulator of hepatic functions, but its role in ALD is mostly unexplored. In this study, we used mice with liver-specific PTP1B disruption as well as pharmacological inhibition to investigate the in vivo function of this phosphatase in ALD. We report upregulation of hepatic PTP1B in the chronic plus binge mouse model and, importantly, in liver biopsies of alcoholic hepatitis patients. Also, mice with hepatic PTP1B disruption attenuated ethanol-induced injury, inflammation, and steatosis compared with ethanol-fed control animals. Moreover, PTP1B deficiency was associated with decreased ethanol-induced oxidative stress in vivo and ex vivo. Further, pharmacological modulation of oxidative balance in hepatocytes identified diminished oxidative stress as a contributor to the salutary effects of PTP1B deficiency. Notably, PTP1B pharmacological inhibition elicited beneficial effects and mitigated hepatic injury, inflammation, and steatosis caused by ethanol feeding. In summary, these findings causally link hepatic PTP1B and ALD and define a potential therapeutic target for the management of this disease.
Graphical abstract
Highlights
-
•
Hepatic PTP1B expression is elevated in a mouse model of ALD and AH patients.
-
•
Liver-specific PTP1B disruption ameliorated ALD in mice.
-
•
PTP1B deficiency was associated with decreased ethanol-induced oxidative stress.
-
•
PTP1B pharmacological inhibition attenuated alcohol-induced hepatic injury in mice.
Related collections
Most cited references60
- Record: found
- Abstract: found
- Article: not found
Mouse model of chronic and binge ethanol feeding (the NIAAA model).
- Record: found
- Abstract: found
- Article: not found
CYP2E1 and oxidative liver injury by alcohol.
- Record: found
- Abstract: found
- Article: not found