- Record: found
- Abstract: found
- Article: found
A novel missense mutation of the NAT10 gene in a juvenile Schnauzer dog with chronic respiratory tract infections
Read this article at
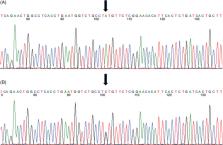
Abstract
An 18‐month‐old intact male Schnauzer dog was evaluated for chronic, lifelong respiratory tract infections that were unresponsive to administration of a variety of antibiotics and corticosteroids. The dog developed persistent vomiting and diarrhea around 1 year of age that was minimally responsive to diet change, antibiotics, and corticosteroids. Despite supportive care, the dog was ultimately euthanized at 20 months of age due to persistent respiratory and gastrointestinal disease. Whole genome sequencing discovered a deleterious missense A/C mutation within the NAT10 gene, a gene essential for microtubule acetylation, appropriate ciliary development, and cytokinesis. Pipeline analysis of the genomes of 579 dogs from 55 breeds did not detect this mutation. Though never described in veterinary medicine, NAT10 mutation occurs in humans with ciliary aplasia, suggesting a pathophysiological mechanism for this dog and highlighting an associated mutation or possible novel genetic cause of chronic respiratory infections in dogs.
Related collections
Most cited references15

- Record: found
- Abstract: found
- Article: found
Trimmomatic: a flexible trimmer for Illumina sequence data

- Record: found
- Abstract: found
- Article: found