- Record: found
- Abstract: found
- Article: not found
Cerebral cavernous malformations arise from endothelial gain of MEKK3-KLF2/4 signaling

Abstract
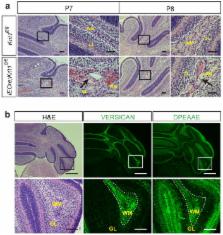
Cerebral cavernous malformations (CCMs) are common inherited and sporadic vascular malformations that cause stroke and seizures in younger individuals 1 . CCMs arise from endothelial cell loss of KRIT1, CCM2, or PDCD10, non-homologous proteins that form an adaptor complex 2 . How disruption of the CCM complex results in disease remains controversial, with numerous signaling pathways (including Rho 3, 4 , SMAD 5 and Wnt/β-catenin 6 ) and processes such as endothelial-mesenchymal transition (EndMT) 5 proposed to play causal roles. CCM2 binds MEKK3 7– 11 , and we have recently demonstrated that CCM complex regulation of MEKK3 is essential during vertebrate heart development 12 . Here, we investigate this mechanism in CCM disease pathogenesis. Using a neonatal mouse model of CCM disease, we find that expression of the MEKK3 target genes KLF2 and KLF4, as well as Rho and ADAMTS protease activity, are increased in the endothelial cells of early CCM lesions. In contrast, we find no evidence of EndMT or increased SMAD or Wnt signaling during early CCM formation. Endothelial-specific loss of Mekk3, Klf2, or Klf4 dramatically prevents lesion formation, reverses the increase in Rho activity, and rescues lethality. Consistent with these findings in mice, we demonstrate that endothelial expression of KLF2 and KLF4 is elevated in human familial and sporadic CCM lesions, and that a disease-causing human CCM2 mutation abrogates MEKK3 interaction without affecting CCM complex formation. These studies identify gain of MEKK3 signaling and KLF2/4 function as causal mechanisms for CCM pathogenesis that may be targeted to develop new CCM therapeutics.
Related collections
Most cited references25
- Record: found
- Abstract: found
- Article: not found
The zinc-finger transcription factor Klf4 is required for terminal differentiation of goblet cells in the colon.

- Record: found
- Abstract: found
- Article: found
Cerebral cavernous malformations proteins inhibit Rho kinase to stabilize vascular integrity
- Record: found
- Abstract: found
- Article: not found