- Record: found
- Abstract: found
- Article: found
Evaluation of P-Glycoprotein Inhibitory Potential Using a Rhodamine 123 Accumulation Assay
Read this article at
Abstract
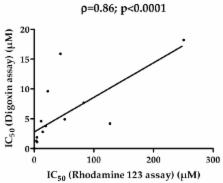
In vitro evaluation of P-glycoprotein (P-gp) inhibitory potential is now a regulatory issue during drug development, in order to predict clinical inhibition of P-gp and subsequent drug–drug interactions. Assays for this purpose, commonly based on P-gp-expressing cell lines and digoxin as a reference P-gp substrate probe, unfortunately exhibit high variability, raising thus the question of developing alternative or complementary tests for measuring inhibition of P-gp activity. In this context, the present study was designed to investigate the use of the fluorescent dye rhodamine 123 as a reference P-gp substrate probe for characterizing P-gp inhibitory potential of 16 structurally-unrelated drugs known to interact with P-gp. 14/16 of these P-gp inhibitors were found to increase rhodamine 123 accumulation in P-gp-overexpressing MCF7R cells, thus allowing the determination of their P-gp inhibitory potential, i.e., their half maximal inhibitor concentration (IC 50) value towards P-gp-mediated transport of the dye. These IC 50 values were in the range of variability of previously reported IC 50 for P-gp and can be used for the prediction of clinical P-gp inhibition according to Food and Drug Administration (FDA) criteria, with notable sensitivity (80%). Therefore, the data demonstrated the feasibility of the use of rhodamine 123 for evaluating the P-gp inhibitory potential of drugs.
Related collections
Most cited references57
- Record: found
- Abstract: found
- Article: not found
Drug-induced mitochondrial dysfunction and cardiotoxicity.
- Record: found
- Abstract: found
- Article: not found