- Record: found
- Abstract: found
- Article: found
Large Scale Characterization of the LC13 TCR and HLA-B8 Structural Landscape in Reaction to 172 Altered Peptide Ligands: A Molecular Dynamics Simulation Study
Read this article at
Abstract
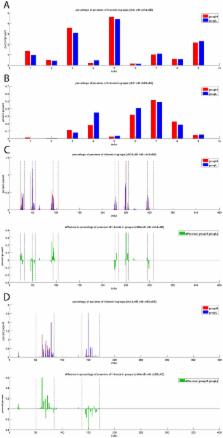
The interplay between T cell receptors (TCRs) and peptides bound by major histocompatibility complexes (MHCs) is one of the most important interactions in the adaptive immune system. Several previous studies have computationally investigated their structural dynamics. On the basis of these simulations several structural and dynamical properties have been proposed as effectors of the immunogenicity. Here we present the results of a large scale Molecular Dynamics simulation study consisting of 100 ns simulations of 172 different complexes. These complexes consisted of all possible point mutations of the Epstein Barr Virus peptide FLRGRAYGL bound by HLA-B*08:01 and presented to the LC13 TCR. We compare the results of these 172 structural simulations with experimental immunogenicity data. We found that simulations with more immunogenic peptides and those with less immunogenic peptides are in fact highly similar and on average only minor differences in the hydrogen binding footprints, interface distances, and the relative orientation between the TCR chains are present. Thus our large scale data analysis shows that many previously suggested dynamical and structural properties of the TCR/peptide/MHC interface are unlikely to be conserved causal factors for peptide immunogenicity.
Author Summary
Immune cells in the human body screen other cells for possible infections. The binding of T-cell receptors (TCR) and parts of pathogens bound by major histocompatibility complexes (MHC) is one of the activation mechanisms of the immune system. There have been many hypotheses as to when such binding will activate the immune system. In this study we performed the, to our knowledge, largest set of Molecular Dynamics simulations of TCR-MHC complexes. We performed 172 simulations each of 100 ns in length. By performing a large number of simulations we obtain insight about which structural features are frequently present in immune system activating and non-activating TCR-MHC complexes. We show that many previously suggested structural features are unlikely to be causal for the activation of the human immune system.
Related collections
Most cited references49
- Record: found
- Abstract: found
- Article: not found
How TCRs bind MHCs, peptides, and coreceptors.
- Record: found
- Abstract: found
- Article: not found
SYFPEITHI: database for MHC ligands and peptide motifs.
- Record: found
- Abstract: found
- Article: not found