- Record: found
- Abstract: found
- Article: found
Green Tea Polyphenols Ameliorate the Early Renal Damage Induced by a High-Fat Diet via Ketogenesis/SIRT3 Pathway
Read this article at
Abstract
Scope
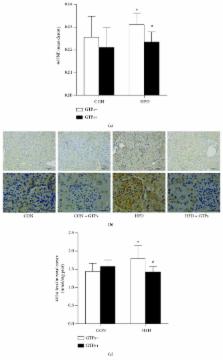
Several reports in the literature have suggested the renoprotective effects of ketone bodies and green tea polyphenols (GTPs). Our previous study found that GTP consumption could elevate the renal expression of the ketogenic rate-limiting enzyme, which was decreased by a high-fat diet (HFD) in rats. Here, we investigated whether ketogenesis can mediate renoprotection by GTPs against an HFD.
Methods and Results
Wistar rats were fed a standard or HFD with or without GTPs for 18 weeks. The renal oxidative stress level, kidney function, renal expression, and activity levels of mitochondrial 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA) synthase 2 (HMGCS2) and sirtuin 3(SIRT3) were detected. The increased renal oxidative stress and the loss of renal function induced by the HFD were ameliorated by GTPs. Renal ketogenesis and SIRT3 expression and activity levels, which were reduced by the HFD, were restored by GTPs. In vitro, HEK293 cells were transfected with the eukaryotic expression plasmid pcDNA HMGCS2. GTP treatment could upregulate HMGCS2 and SIRT3 expression. Although SIRT3 expression was not affected by HMGCS2 transfection, the 4-hydroxy-2-nonenal (4-HNE) level and the acetyl-MnSOD (K122)/MnSOD ratio were reduced in HMGCS2-transfected cells in the context of H 2O 2.
Related collections
Most cited references37
- Record: found
- Abstract: found
- Article: not found
Nutrient-sensitive mitochondrial NAD+ levels dictate cell survival.
- Record: found
- Abstract: found
- Article: not found
PGC1α-dependent NAD biosynthesis links oxidative metabolism to renal protection
- Record: found
- Abstract: found
- Article: not found