- Record: found
- Abstract: found
- Article: found
Experience and Perspectives in the US on the Evolving Treatment Landscape in Spinal Muscular Atrophy
Read this article at
Abstract
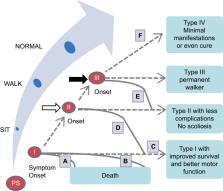
Spinal muscular atrophy (SMA) is a rare, progressive neuromuscular disorder that, until recently, was the most common inherited cause of infant mortality. Since 2016, three disease-modifying therapies have emerged, nusinersen, onasemnogene abeparvovec-xioi, and risdiplam, leading to a transformation in the SMA treatment landscape, changes in disease trajectories, and a profound impact on clinical care. This environment poses a challenge to making informed treatment decisions, including initial treatment choice, treatment changes, and potential use of combination therapies as new data emerge. To better understand factors that influence physician-patient decision-making, a roundtable discussion was convened by Biogen (sponsor) with a panel of four US SMA experts. This report shares the panel’s opinions and clinical experiences, with the goals of helping clinicians and people with SMA and their families to better understand the factors influencing real-world treatment decisions and stimulating a broader discussion in the SMA community. The panelists highlighted that patients are often heavily involved in treatment decisions, and physicians must be aware of current data to guide patients in making the best decisions. Thus, in the absence of data from head-to-head treatment comparisons, physicians’ roles include reviewing treatment options and describing what is known of the benefits, challenges, and potential side effects of each therapy with patients and families. For infants and young children, the panelists expressed a sense of urgency for early intervention to minimize motor function loss, whereas the goal for adults is long-term disease stabilization. In the panelists’ experience, factors that influence patients’ decisions to change to an alternative therapy include convenience, administration route, novelty of therapy, and hope for improved function, while reasons for returning to a previous therapy include a perception of decreased efficacy and side effects. Ongoing clinical trials and analyses of real-world experiences should further inform treatment decisions and optimize patient outcomes.
Graphical Abstract

Related collections
Most cited references58
- Record: found
- Abstract: found
- Article: not found
Nusinersen versus Sham Control in Infantile-Onset Spinal Muscular Atrophy
- Record: found
- Abstract: found
- Article: not found
Nusinersen versus Sham Control in Later-Onset Spinal Muscular Atrophy
- Record: found
- Abstract: found
- Article: not found