- Record: found
- Abstract: found
- Article: found
Microbial Metabolic Potential of Phenol Degradation in Wastewater Treatment Plant of Crude Oil Refinery: Analysis of Metagenomes and Characterization of Isolates
Read this article at
Abstract
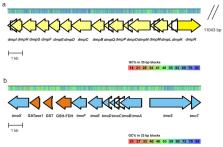
The drilling, processing and transportation of oil are the main sources of pollution in water and soil. The current work analyzes the microbial diversity and aromatic compounds degradation potential in the metagenomes of communities in the wastewater treatment plant (WWTP) of a crude oil refinery. By focusing on the degradation of phenol, we observed the involvement of diverse indigenous microbial communities at different steps of the WWTP. The anaerobic bacterial and archaeal genera were replaced by aerobic and facultative anaerobic bacteria through the biological treatment processes. The phyla Proteobacteria, Bacteroidetes and Planctomycetes were dominating at different stages of the treatment. Most of the established protein sequences of the phenol degradation key enzymes belonged to bacteria from the class Alphaproteobacteria. From 35 isolated strains, 14 were able to grow on aromatic compounds, whereas several phenolic compound-degrading strains also degraded aliphatic hydrocarbons. Two strains, Acinetobacter venetianus ICP1 and Pseudomonas oleovorans ICTN13, were able to degrade various aromatic and aliphatic pollutants and were further characterized by whole genome sequencing and cultivation experiments in the presence of phenol to ascertain their metabolic capacity in phenol degradation. When grown alone, the intermediates of catechol degradation, the meta or ortho pathways, accumulated into the growth environment of these strains. In the mixed cultures of the strains ICP1 and ICTN13, phenol was degraded via cooperation, in which the strain ICP1 was responsible for the adherence of cells and ICTN13 diminished the accumulation of toxic intermediates.
Related collections
Most cited references76
- Record: found
- Abstract: found
- Article: not found
ProtTest: selection of best-fit models of protein evolution.
- Record: found
- Abstract: found
- Article: not found
METAXA2: improved identification and taxonomic classification of small and large subunit rRNA in metagenomic data.
- Record: found
- Abstract: found
- Article: not found