- Record: found
- Abstract: found
- Article: not found
Structure-Based Design, Synthesis and Validation of CD4-Mimetic Small Molecule Inhibitors of HIV-1 Entry: Conversion of a Viral Entry Agonist to an Antagonist

Read this article at

Conspectus
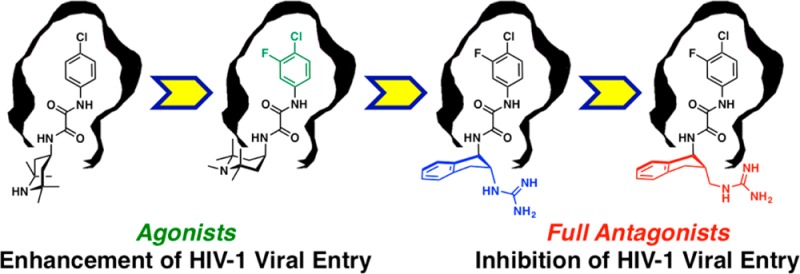
This Account provides an overview of a multidisciplinary consortium focused on structure-based strategies to devise small molecule antagonists of HIV-1 entry into human T-cells, which if successful would hold considerable promise for the development of prophylactic modalities to prevent HIV transmission and thereby alter the course of the AIDS pandemic.
Entry of the human immunodeficiency virus (HIV) into target T-cells entails an interaction between CD4 on the host T-cell and gp120, a component of the trimeric envelope glycoprotein spike on the virion surface. The resultant interaction initiates a series of conformational changes within the envelope spike that permits binding to a chemokine receptor, formation of the gp41 fusion complex, and cell entry. A hydrophobic cavity at the CD4–gp120 interface, defined by X-ray crystallography, provided an initial site for small molecule antagonist design. This site however has evolved to facilitate viral entry. As such, the binding of prospective small molecule inhibitors within this gp120 cavity can inadvertently trigger an allosteric entry signal.
Structural characterization of the CD4–gp120 interface, which provided the foundation for small molecule structure-based inhibitor design, will be presented first. An integrated approach combining biochemical, virological, structural, computational, and synthetic studies, along with a detailed analysis of ligand binding energetics, revealed that modestly active small molecule inhibitors of HIV entry can also promote viral entry into cells lacking the CD4 receptor protein; these competitive inhibitors were termed small molecule CD4 mimetics. Related congeners were subsequently identified with both improved binding affinity and more potent viral entry inhibition. Further assessment of the affinity-enhanced small molecule CD4 mimetics demonstrated that premature initiation of conformational change within the viral envelope spike, prior to cell encounter, can lead to irreversible deactivation of viral entry machinery. Related congeners, which bind the same gp120 site, possess different propensities to elicit the allosteric response that underlies the undesired enhancement of CD4-independent viral entry.
Subsequently, key hotspots in the CD4–gp120 interface were categorized using mutagenesis and isothermal titration calorimetry according to the capacity to increase binding affinity without triggering the allosteric signal. This analysis, combined with cocrystal structures of small molecule viral entry agonists with gp120, led to the development of fully functional antagonists of HIV-1 entry. Additional structure-based design exploiting two hotspots followed by synthesis has now yielded low micromolar inhibitors of viral entry.
Related collections
Most cited references28
- Record: found
- Abstract: found
- Article: not found
The HIV-1 envelope glycoproteins: fusogens, antigens, and immunogens.
- Record: found
- Abstract: found
- Article: not found
HIV-1 evades antibody-mediated neutralization through conformational masking of receptor-binding sites.
- Record: found
- Abstract: found
- Article: not found