- Record: found
- Abstract: found
- Article: found
Dimerisation of the PICTS complex via LC8/Cut-up drives co-transcriptional transposon silencing in Drosophila
Read this article at
Abstract
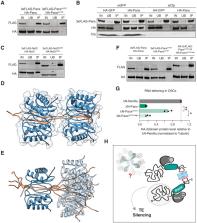
In animal gonads, the PIWI-interacting RNA (piRNA) pathway guards genome integrity in part through the co-transcriptional gene silencing of transposon insertions. In Drosophila ovaries, piRNA-loaded Piwi detects nascent transposon transcripts and instructs heterochromatin formation through the Panoramix- induced co- transcriptional silencing (PICTS) complex, containing Panoramix, Nxf2 and Nxt1. Here, we report that the highly conserved dynein light chain LC8/Cut-up (Ctp) is an essential component of the PICTS complex. Loss of Ctp results in transposon de-repression and a reduction in repressive chromatin marks specifically at transposon loci. In turn, Ctp can enforce transcriptional silencing when artificially recruited to RNA and DNA reporters. We show that Ctp drives dimerisation of the PICTS complex through its interaction with conserved motifs within Panoramix. Artificial dimerisation of Panoramix bypasses the necessity for its interaction with Ctp, demonstrating that conscription of a protein from a ubiquitous cellular machinery has fulfilled a fundamental requirement for a transposon silencing complex.
Related collections
Most cited references85
- Record: found
- Abstract: found
- Article: not found
Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method.

- Record: found
- Abstract: found
- Article: found
Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2
- Record: found
- Abstract: found
- Article: not found