- Record: found
- Abstract: found
- Article: found
Identification of the Biomarkers and Pathological Process of Heterotopic Ossification: Weighted Gene Co-Expression Network Analysis
Read this article at
Abstract
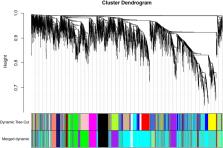
Heterotopic ossification (HO) is the formation of abnormal mature lamellar bone in extra-skeletal sites, including soft tissues and joints, which result in high rates of disability. The understanding of the mechanism of HO is insufficient. The aim of this study was to explore biomarkers and pathological processes in HO+ samples. The gene expression profile GSE94683 was downloaded from the Gene Expression Omnibus database. Sixteen samples from nine HO- and seven HO+ subjects were analyzed. After data preprocessing, 3,529 genes were obtained for weighted gene co-expression network analysis. Highly correlated genes were divided into 13 modules. Finally, the cyan and purple modules were selected for further study. Gene ontology functional annotation and Kyoto Encyclopedia of Genes and Genomes pathway enrichment indicated that the cyan module was enriched in a variety of components, including protein binding, membrane, nucleoplasm, cytosol, poly(A) RNA binding, biosynthesis of antibiotics, carbon metabolism, endocytosis, citrate cycle, and metabolic pathways. In addition, the purple module was enriched in cytosol, mitochondrion, protein binding, structural constituent of ribosome, rRNA processing, oxidative phosphorylation, ribosome, and non-alcoholic fatty liver disease. Finally, 10 hub genes in the cyan module [actin related protein 3 ( ACTR3), ADP ribosylation factor 4 ( ARF4), progesterone receptor membrane component 1 ( PGRMC1), ribosomal protein S23 ( RPS23), mannose-6-phosphate receptor ( M6PR), WD repeat domain 12 ( WDR12), synaptosome associated protein 23 ( SNAP23), actin related protein 2 ( ACTR2), siah E3 ubiquitin protein ligase 1 ( SIAH1), and glomulin ( GLMN)] and 2 hub genes in the purple module [proteasome 20S subunit alpha 3 ( PSMA3) and ribosomal protein S27 like ( RPS27L)] were identified. Hub genes were validated through quantitative real-time polymerase chain reaction. In summary, 12 hub genes were identified in two modules that were associated with HO. These hub genes could provide new biomarkers, therapeutic ideas, and targets in HO.
Related collections
Most cited references67
- Record: found
- Abstract: found
- Article: not found
Cytoscape: a software environment for integrated models of biomolecular interaction networks.

- Record: found
- Abstract: found
- Article: found
WGCNA: an R package for weighted correlation network analysis
- Record: found
- Abstract: found
- Article: not found