- Record: found
- Abstract: found
- Article: found
Importance of serial changes in biomarkers in idiopathic pulmonary fibrosis
Read this article at
Abstract
The clinical significance of serial changes in serum biomarkers in patients with idiopathic pulmonary fibrosis (IPF) remains to be established. This retrospective study was conducted to clarify the associations of serial changes in serum Krebs von den Lungen-6 (KL-6) and surfactant protein-D (SP-D) with changes in physiological indices and overall mortality in IPF.
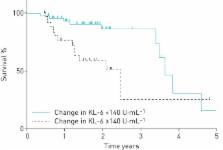
The study subjects were 75 patients with IPF. The 6 month change in serum KL-6 was significantly correlated with changes in the percentage of the predicted forced vital capacity (FVC % pred) and the percentage of the predicted diffusing capacity of the lung for carbon monoxide (% D LCO), while the 6 month change in serum SP-D was correlated only with % D LCO. During the mean follow-up period of 647 days, 22 (29.3%) patients died. An increase in serum KL-6 over a 6 month period was a significant predictor of mortality even after adjustment for %FVC, % D LCO and serum KL-6 at the baseline (hazard ratio 1.10 per 100 U·mL −1, 95% CI 1.01–1.18, p=0.03), whereas the 6 month increase in serum SP-D was not significant.
Serial measurements of serum KL-6 may provide additional prognostic information compared to that provided by physiological parameters in patients with IPF.
Abstract
Serial changes in serum KL-6 are associated with changes in physiological variables and can predict survival in IPF http://ow.ly/hCkb30eauLg
Related collections
Most cited references33
- Record: found
- Abstract: found
- Article: not found
A multidimensional index and staging system for idiopathic pulmonary fibrosis.
- Record: found
- Abstract: found
- Article: not found
Pirfenidone for idiopathic pulmonary fibrosis: analysis of pooled data from three multinational phase 3 trials
- Record: found
- Abstract: found
- Article: not found