- Record: found
- Abstract: found
- Article: found
Whole genome amplification with SurePlex results in better copy number alteration detection using sequencing data compared to the MALBAC method
Read this article at
Abstract
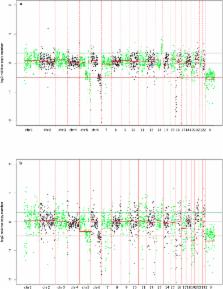
Current whole genome amplification (WGA) methods lead to amplification bias resulting in over- and under-represented regions in the genome. Nevertheless, certain WGA methods, such as SurePlex and subsequent arrayCGH analysis, make it possible to detect copy number alterations (CNAs) at a 10 Mb resolution. A more uniform WGA combined with massive parallel sequencing (MPS), however, could allow detection at higher resolution and lower cost. Recently, MALBAC, a new WGA method, claims unparalleled performance. Here, we compared the well-established SurePlex and MALBAC WGA for their ability to detect CNAs in MPS generated data and, in addition, compared PCR-free MPS library preparation with the standard enrichment PCR library preparation. Results showed that SurePlex amplification led to more uniformity across the genome, allowing for a better CNA detection with less false positives compared to MALBAC amplified samples. An even more uniform coverage was observed in samples following a PCR-free library preparation. In general, the combination of SurePlex and MPS led to the same chromosomal profile compared to a reference arrayCGH from unamplified genomic DNA, underlining the large potential of MPS techniques in CNA detection from a limited number of DNA material.
Related collections
Most cited references15
- Record: found
- Abstract: found
- Article: not found
Whole genome amplification from a single cell: implications for genetic analysis.

- Record: found
- Abstract: found
- Article: found
A Quantitative Comparison of Single-Cell Whole Genome Amplification Methods

- Record: found
- Abstract: found
- Article: found