- Record: found
- Abstract: found
- Article: found
Modulation of Host Angiogenesis as a Microbial Survival Strategy and Therapeutic Target
review-article
14 April 2016
Read this article at
There is no author summary for this article yet. Authors can add summaries to their articles on ScienceOpen to make them more accessible to a non-specialist audience.
Abstract
Angiogenesis in Health and Disease
The blood vascular system is essential to the development and maintenance of tissues
of multicellular eukaryotes. Its roles include internal transport and delivery of
oxygen and nutrients and immune surveillance and trafficking of cells and molecules
of the innate immune system to sites of tissue damage. Formation of blood vessels
occurs by two principal modes: assembly of endothelial progenitor cells into vascular
networks (vasculogenesis), which takes place predominantly in embryonic life, and
the expansion of existing vascular systems by sprouting of new blood vessels from
existing ones (angiogenesis) [1]. Angiogenesis is a sequential process guided by angiogenic
cues, notably, hypoxia inducible factor (HIF)-1, vascular endothelial growth factor
(VEGF), fibroblast growth factor (FGF), angiopoietin-2, and chemokines released by
hypoxic, inflammatory, or neoplastic cells [1].
Whereas physiological angiogenesis is essential for normal tissue growth, remodeling,
and regeneration, dysregulated angiogenesis plays a pivotal role in disease states,
including cancer, inflammatory diseases, atherosclerosis, and diabetic retinopathy
[2]. Importantly, cancer cells can exploit angiogenesis to support their own proliferation
and metastatic dissemination. This so-called tumor angiogenesis has been the focus
of intense research, leading to the discovery of a novel class of antineoplastic drugs
[3]. During infection, angiogenesis is induced when microbial motifs are detected
in concert with damage-associated molecular patterns. Specifically, bacterial ligands
such as LPS and unmethylated CpG activate mammalian Toll-like receptors (TLRs) 2,
4, 7, and 9, while adenosine, a danger signal that accumulates rapidly in ischemic
or damaged tissues, synergizes with TLRs to induce the synthesis and release of VEGF
and recruitment of endothelial progenitor cells [4]. The ensuing inflammatory angiogenic
response facilitates the migration of leukocytes to infected tissue and wound repair.
Moreover, an emerging concept links angiogenesis to innate immunity, implying that
an adequate angiogenic response is required for control and clearance of invading
pathogens [5–7]. Intriguingly, some microbial pathogens manipulate the host angiogenic
response, either suppressing it to enhance their persistence in tissues or hijacking
angiogenesis for their own ends. Deciphering such interactions may uncover new therapeutic
targets for some of the most tenacious infectious diseases. In this mini-review, we
highlight examples where modulation of host angiogenesis has been shown to play an
important role in microbial pathogenesis.
Regional Events at Infection Sites Control Microbial Sequestration and Killing
The evolution of a circulatory system enables a systemic immune response but opens
the way for rapid dissemination of pathogens within the host. Rapid microbial dissemination
is controlled via early local events that wall off invading pathogens [8]. Microbial
sequestration addresses contrasting needs; it must enable migration of immune cells
and antimicrobial molecules into infected tissue while preventing pathogens from gaining
access to the circulatory system. Failure to achieve these goals results in microbial
persistence or dissemination, respectively. The early events that occur within hours
of microbial invasion include triggering of the complement cascade and platelet aggregation
followed by the expression of adhesion molecules (endothelial-leukocyte adhesion molecule
[ELAM]-1, intercellular adhesion molecule [ICAM]-1, and vascular cell adhesion molecule
[VCAM]-1) on activated endothelial cells, facilitating the influx of immune cells
to infected tissue [9].
The microenvironmental conditions at the site of infection are characterized by low
oxygen pressure and high concentrations of lactate and reductive metabolites. This
is especially true if the local vasculature is directly disrupted by infection. The
heterodimeric transcription factor HIF-1 is the pivotal regulator of angiogenesis
and myeloid cell function under hypoxic conditions. HIF-1α levels are dynamically
controlled by oxygen-dependent prolyl hydroxylase domain (PHD) proteins that regulate
HIF stability [10]. Moreover, HIF-1 and NF-kB signaling are strongly interdependent,
with intact NF-kB signaling shown to be required for hypoxic HIF-1 induction [11,12].
HIF-1 activation is observed in infections with bacteria, viruses, fungi, and protozoa
[13]. Interestingly, hypoxia-independent activation of HIF-1α is induced by iron deprivation,
suggesting that bacterial siderophores may also trigger this pathway [14]. Myeloid
aggregation, motility, invasion, and bacterial killing are critically dependent on
HIF-1α, which allows myeloid cells to function under conditions of low oxygen pressure
by switching to glycolytic metabolism [15]. In sum, HIF-1 activation of VEGF signaling
and angiogenesis likely act in concert with myeloid cell activation and trafficking
to keep tissue-invasive pathogens in check.
Some Pathogens Enhance Host Angiogenesis to Support Infection
Infection-associated angiogenesis has been described in diverse infections caused
by bacteria, viruses, protozoa, and fungi (Table 1). Conceptually, infectious angiogenesis
may be classified as either direct induction of host angiogenesis by pathogen-derived
molecules or angiogenesis driven by a nonspecific host inflammatory response. Both
Bartonella henselae and Kaposi sarcoma-associated herpesvirus (KSHV) induce rampant
angiogenesis, resulting in severe illness in persons with deficient cellular immunity,
such as patients with AIDS. The B. henselae adhesin A (BadA) and type IV secretion
system VirB/D4 mediate bacterial endothelial cell adherence and uptake followed by
activation of a proangiogenic phenotype, thereby expanding the host cell habitat of
this pathogen [16]. KSHV expresses several factors that either directly activate the
formation of blood vessels (viral interleukin 6 [vIL-6], vCCL-1, and vCCL-II) or indirectly
activate cell pathways, leading to angiogenesis (vGPCR, vFLIP, K1, K15, KSHV miRNAs)
[17]. Virus-driven angiogenesis enables propagation of KSHV by recruiting uninfected
endothelial lineage and hematopoietic cells for further infection and reactivation
of KSHV in latently infected cells [18].
10.1371/journal.ppat.1005479.t001
Table 1
Notable pathogens associated with modulation of host angiogenesis.
Pathogens associated with pro-angiogenesis
Mechanisms discovered
References
Bartonella henselae
Reprogramming of human myeloid cells towards a tumor-associated macrophage–like proangiogenic
phenotype.
[32]
Bartonella adhesin A (BadA) mediates binding to fibronectin, adherence to endothelial
cells, and secretion of VEGF.
[16]
The type IV secretion system VirB/D4 translocates several Bartonella effector proteins
(Beps) into the cytoplasm of infected endothelial cells, resulting in uptake of bacterial
aggregates, inhibition of apoptosis, and activation of a proangiogenic phenotype.
[33]
Mycobacterium tuberculosis
Mycobacteria induce abnormal leaky granuloma-associated angiogenesis, which promotes
mycobacterial growth and increases spread of infection to new tissue sites.
[6,19]
Candida albicans
C. albicans stimulates vascularization in infected brain and kidney abscesses and
activates endothelial cell genes involved in chemotaxis and angiogenesis.
[34,35]
Kaposi Sarcoma Herpesvirus (KSHV)
KSHV expresses molecules that directly activate the formation of blood vessels: viral
interleukin 6 (vIL-6), vCCL-1, vCCL-II, vGPCR, vFLIP, K1, K15, and KSHV miRNAs.
[17,18]
Cytomegalovirus (CMV)
CMV-secreted pUL7 carcinoembryonic antigen-related cell adhesion molecule (CEACAM)–related
protein induces angiogenesis in endothelial cells via STAT3/ERK1/2 activation and
IL-6 secretion.
[36]
Hepatitis C virus (HCV)
HCV-mediates hepatic angiogenesis by stabilizing cellular HIF-1α via the NF-κB pathway
to up-regulate VEGF and other proangiogenic factors.
[37]
Human papillomavirus (HPV)
HPV E6 protein inhibits p53 and stabilizes HIF-1α to up-regulate VEGF, favoring formation
of new blood vessels and increasing permeability of existing blood vessels.
[38]
Schistosoma mansonii
S. mansonii soluble egg metabolites induce hepatic neovascularization by up-regulating
endothelial cell VEGF as well as directly inducing endothelial cell proliferation,
migration, and sprouting.
[39–41]
Pathogens associated with inhibition of angiogenesis
Bacillus anthracis
Bacillus anthracis protective antigen (PA) inhibits VEGF and basic fibroblast growth
factor (bFGF)-induced endothelial cell angiogenesis.
[42]
Pseudomonas aeruginosa
P. aeruginosa hemolytic phospholipase C at picomolar concentrations is selectively
lethal to endothelial cells and inhibits angiogenesis.
[43]
Aspergillus fumigatus
Down-regulation of HIF-1α, VEGF-A, bFGF, and VEGF receptors 1 and 2 is dependent on
A. fumigatus secondary metabolism under the transcriptional regulation of LaeA.
[7,24]
M. tuberculosis, the causative agent of tuberculosis, has not been found to produce
bacterial angiogenic factors, yet its ability to survive and persist in the host is
intimately related to pathological angiogenesis [6,19]. M. tuberculosis elicits the
formation of dense cellular aggregates (granulomas) that wall off the pathogen. The
presence of viable mycobacteria within macrophages in granulomas triggers VEGF-dependent
tumor–like angiogenesis associated with dysfunctional (leaky) blood vessels. Dysregulated
angiogenesis further limits perfusion of the granuloma core, exacerbating hypoxia
and causing caseating necrosis, a hallmark of this infection (Fig 1). Pathological
angiogenesis may be an important cause of inadequate delivery of antibiotic drugs
and immune cells to the center of the granuloma, necessitating multidrug combinations
and protracted treatment courses to eradicate the disease [6,19].
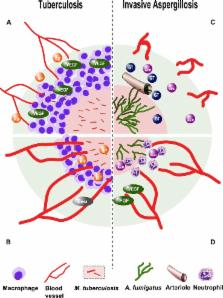
10.1371/journal.ppat.1005479.g001
Fig 1
Modulation of angiogenesis in tuberculosis and invasive aspergillosis.
A. Vascular endothelial growth factor (VEGF)-mediated, host-induced pathological angiogenesis
in M. tuberculosis granulomas restricts perfusion of the granuloma core and attenuates
antituberculosis drug efficacy of rifampicin (RIF). B. Treatment with the angiogenesis-inhibiting
drug bevacizumab (Beva) reverses pathological angiogenesis, enhances perfusion of
the granuloma core, and synergizes with rifampicin. C. A. fumigatus hyphae invade
pulmonary arterioles and induce intravascular thrombosis. The compensatory angiogenic
response is down-regulated by gliotoxin (GT) and other fungal secondary metabolites,
further limiting perfusion of infected tissue with the antifungal drug amphotericin
B (AmB). D. Treatment with proangiogenic growth factors VEGF and fibroblast growth
factor (FGF) counteracts the action of gliotoxin and enhances the influx of polymorphonuclear
leukocytes and antifungal drugs to the site of infection.
Attenuation of Host Angiogenesis Creates Sequestered Niches Where Pathogens Persist
Inhibition of angiogenesis during infection interferes with tissue healing and facilitates
a hypoxic and/or necrotic milieu that compromises immune function and favors pathogen
persistence (Table 1). A. fumigatus produces life-threatening pulmonary infection
in immunocompromised individuals, principally patients with hematological malignancies
and recipients of hematopoietic stem cell transplantation [20]. In the setting of
profound neutropenia, airborne spores (conidia) are inhaled into pulmonary alveoli,
where they germinate and form tissue-invasive filaments (hyphae) that bore through
the alveolar–capillary barrier and invade pulmonary arterioles [20]. Angioinvasion
is associated with endothelial injury, tissue factor expression, triggering of the
coagulation cascade, and platelet activation [21]. Collectively, these processes impair
vascular perfusion of Aspergillus-infected lung tissue, producing a necrotic core
where fungal hyphae proliferate abundantly, surrounded by a peripheral zone of alveolar
hemorrhage (Fig 1) [22]. The importance of adaptation to hypoxia for A. fumigatus
pathogenesis is underscored by work showing that deletion of the SrbA gene, which
is essential for survival in hypoxic environments, renders A. fumigatus nonvirulent
[23]. Invasive pulmonary aspergillosis is associated with a rapid increase in tumor
necrosis factor (TNF)α transcription in mouse lungs but down-regulation of angiogenesis
mediators that are normally induced by this cytokine: VEGF, FGF, and their receptors
[24]. Uncoupling of inflammatory mediators and angiogenesis is further evident in
reduced microvascular density around necrotic pulmonary lesions [7,25]. Inhibition
of angiogenesis is mediated by A. fumigatus secondary metabolites, chiefly gliotoxin,
under the transcriptional control of LaeA [24]. Attenuated angiogenesis likely perpetuates
tissue hypoxia and limits trafficking of immune cells and antifungal drugs into the
site of Aspergillus infection [5,20]. Thus, the vasculopathy of invasive aspergillosis
plays a pathogenic role by restricting innate immune cell traffic to the site of infection
and optimizing local growth conditions for the fungus.
Modulation of Host Angiogenesis as a Therapeutic Target in Infections
The concept of angiogenesis modulation as a novel microbial virulence factor suggests
the potential for attenuating pathogenicity using vascular-active molecules. Cancer
research has produced numerous monoclonal antibodies and small molecules that target
VEGF and its receptor (VEGFR) [3,26,27]. Originally thought to deprive tumors of their
vascular supply, these agents are now believed to increase perfusion and alleviate
hypoxia by normalizing tumor vasculature [27]. Similarly, angiogenesis modulators
have little if any direct antimicrobial activity but act synergistically with conventional
antimicrobials by enhancing drug delivery to the anatomical site of infection.
This idea has been explored in a rabbit model of M. tuberculosis infection and a zebrafish
model of Mycobacterium marinum infection [6,19]. Inhibition of angiogenesis using
bevacizumab, an anti-VEGF-A monoclonal antibody [6], and VEGFR tyrosine kinase inhibitors
SU5416 and pazopanib [19] prevented the formation of abnormal ectopic blood vessels
around mycobacterial granulomas, improved granuloma perfusion, and decreased necrotic
tissue volume, bacterial burden, and dissemination without directly affecting mycobacterial
growth in vitro (Fig 1) [6,19]. Moreover, pazopanib treatment alone significantly
increased survival in the M. marinum zebrafish model, and SU5416 potentiated the activity
of the first-line antituberculosis drug rifampicin [19].
In contrast, the vasculopathy of invasive aspergillosis is reversed following repletion
of proangiogenic factors [7]. Treatment with VEGF and FGF alone significantly increased
survival in a neutropenic mouse model of invasive pulmonary aspergillosis, and both
growth factors acted synergistically with the antifungal drug amphotericin B to enhance
survival and decrease pulmonary fungal burden (Fig 1) [7]. FGF enhanced the generation
of CD31-positive vessels and was associated with neutrophil infiltrates around A.
fumigatus infection sites. Interestingly, FGF had a more potent effect on mouse survival
and fungal burden than did VEGF, a fact consistent with the association of VEGF with
immature and hyperpermeable blood vessels [7].
These preliminary findings should be viewed within the context of the grand challenges
to healthcare presented by M. tuberculosis and A. fumigatus [28,29]. M. tuberculosis
infects one-third of the world’s population and is the second greatest cause of infectious
mortality worldwide [29]. Currently, treatment involves complex multidrug regimens
lasting months, which many patients do not tolerate. Moreover, extensive resistance
to antituberculosis drugs has emerged in some parts of the world [29]. Invasive aspergillosis
is lethal in about one-third of patients [30], and resistance to voriconazole, the
foremost drug used to treat this infection, is spreading across Europe and Asia [31].
Vascular targeted therapies may herald the prospect of more effective antimicrobial
drug delivery, allowing shorter, simpler treatment regimens and more efficient pathogen
clearance.
Related collections
Most cited references31
- Record: found
- Abstract: found
- Article: not found
Angiogenesis in life, disease and medicine.
Peter Carmeliet (2005)
- Record: found
- Abstract: found
- Article: not found
HIF-1alpha is essential for myeloid cell-mediated inflammation.
Thorsten Cramer, Yuji Yamanishi, Björn E Clausen … (2003)
- Record: found
- Abstract: not found
- Article: not found
Tuberculosis.
Alimuddin Zumla, Mario Raviglione, Richard Hafner … (2013)