- Record: found
- Abstract: found
- Article: not found
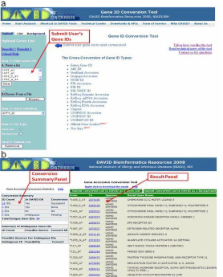
DAVID gene ID conversion tool
Read this article at

Abstract
Our current biological knowledge is spread over many independent bioinformatics databases where many different types of gene and protein identifiers are used. The heterogeneous and redundant nature of these identifiers limits data analysis across different bioinformatics resources. It is an even more serious bottleneck of data analysis for larger datasets, such as gene lists derived from microarray and proteomic experiments. The DAVID Gene ID Conversion Tool (DICT), a web-based application, is able to convert user's input gene or gene product identifiers from one type to another in a more comprehensive and high-throughput manner with a uniquely enhanced ID-ID mapping database.
Related collections
Most cited references9
- Record: found
- Abstract: found
- Article: not found
DAVID: Database for Annotation, Visualization, and Integrated Discovery.
- Record: found
- Abstract: found
- Article: not found
Entrez Gene: gene-centered information at NCBI
- Record: found
- Abstract: found
- Article: not found