- Record: found
- Abstract: found
- Article: found
Correction of diverse muscular dystrophy mutations in human engineered heart muscle by single-site genome editing
Read this article at
Abstract
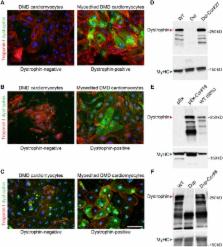
CRISPR-mediated genome editing efficiently corrected cardiac abnormalities associated with DMD in human engineered heart tissue.
Abstract
Genome editing with CRISPR/Cas9 is a promising new approach for correcting or mitigating disease-causing mutations. Duchenne muscular dystrophy (DMD) is associated with lethal degeneration of cardiac and skeletal muscle caused by more than 3000 different mutations in the X-linked dystrophin gene ( DMD). Most of these mutations are clustered in “hotspots.” There is a fortuitous correspondence between the eukaryotic splice acceptor and splice donor sequences and the protospacer adjacent motif sequences that govern prokaryotic CRISPR/Cas9 target gene recognition and cleavage. Taking advantage of this correspondence, we screened for optimal guide RNAs capable of introducing insertion/deletion (indel) mutations by nonhomologous end joining that abolish conserved RNA splice sites in 12 exons that potentially allow skipping of the most common mutant or out-of-frame DMD exons within or nearby mutational hotspots. We refer to the correction of DMD mutations by exon skipping as myoediting. In proof-of-concept studies, we performed myoediting in representative induced pluripotent stem cells from multiple patients with large deletions, point mutations, or duplications within the DMD gene and efficiently restored dystrophin protein expression in derivative cardiomyocytes. In three-dimensional engineered heart muscle (EHM), myoediting of DMD mutations restored dystrophin expression and the corresponding mechanical force of contraction. Correcting only a subset of cardiomyocytes (30 to 50%) was sufficient to rescue the mutant EHM phenotype to near-normal control levels. We conclude that abolishing conserved RNA splicing acceptor/donor sites and directing the splicing machinery to skip mutant or out-of-frame exons through myoediting allow correction of the cardiac abnormalities associated with DMD by eliminating the underlying genetic basis of the disease.
Related collections
Most cited references20
- Record: found
- Abstract: found
- Article: not found
Efficient genome modification by CRISPR-Cas9 nickase with minimal off-target effects.
- Record: found
- Abstract: found
- Article: not found
Evidence-based path to newborn screening for Duchenne muscular dystrophy.

- Record: found
- Abstract: found
- Article: found