- Record: found
- Abstract: found
- Article: not found
Diseases of the Liver and Biliary Tract
chapter-article
There is no author summary for this article yet. Authors can add summaries to their articles on ScienceOpen to make them more accessible to a non-specialist audience.
Abstract
DIAGNOSTIC STRATEGY FOR LIVER DISEASE
The liver has many diverse functions related to hepatic blood flow; protein, carbohydrate,
and fat metabolism; detoxification and excretion of drugs and toxins; and formation
and elimination of bile. Consequently, the clinical and laboratory abnormalities associated
with liver failure are diverse.
Overview of the Diagnostic Strategy
•
Clinical, laboratory, and imaging studies to identify the presence of liver disease
•
Characterization of the functional aspects of the hepatic disease
•
Determination of an etiologic or histologic diagnosis, which usually requires a liver
biopsy
Acute versus Chronic Disease
The clinical approach and management of patients with hepatic disease is dictated
largely by the acute versus chronic nature of the hepatic disorder. Historical, physical,
laboratory, and radiographic findings may suggest whether the hepatic disease is acute
or chronic, but hepatic biopsy often is required for definitive evaluation. Classifying
a disorder as acute or chronic has diagnostic, therapeutic, and prognostic implications.
•
In acute hepatic failure, toxic or infectious causes are common, and intensive supportive
care is warranted to allow time for hepatic regeneration. The long-term prognosis
for recovery is favorable if the animal survives the initial stages.
•
Chronic hepatic disorders are more likely to be accompanied by irreversible changes
(cirrhosis); thus, the long-term prognosis may not be favorable.
Clinical Signs
Clinical signs of liver disease include those typically associated with hepatic dysfunction,
such as jaundice, hepatic encephalopathy (HE), ascites, and excessive bleeding, and
nonspecific signs such as vomiting, diarrhea, anorexia, lethargy, and weight loss,
which overlap with signs of other body system disorders.
Nonspecific Signs
•
Vomiting is a common sign of liver disease. Hematemesis suggests gastroduodenal ulceration,
a recognized complication of hepatobiliary disease.
•
Diarrhea occurs less frequently than vomiting and is characteristically small-bowel
diarrhea.
•
Anorexia is a common but nonspecific sign of hepatobiliary disease.
•
Weight loss and stunted growth are nonspecific signs that suggest chronic rather than
acute hepatic disease.
Polyuria and Polydipsia
•
Polyuria and polydipsia (PU/PD) may be important presenting clinical signs in dogs
with liver disease. The mechanism is multifactorial and includes psychogenic polydipsia,
hypercortisolism, and renal concentrating defects.
Signs of Abnormal Bilirubin Excretion
•
Pigmented urine (bilirubinuria) and jaundice (icterus) of the sclera, oral mucous
membranes, and skin are classic signs of cholestatic liver disease. However, these
findings are not specific for hepatobiliary disease and can also be caused by hemolytic
disorders.
•
Acholic (gray) feces occur secondary to severe cholestasis (usually from common bile
duct obstruction), which prevents bilirubin in the bile from entering the intestinal
tract and imparting the normal brown color to the feces.
Coagulopathy
•
Excessive bleeding (i.e., hemorrhages of the skin and mucous membranes, melena, and
hematuria) occasionally is associated with liver disease, especially if hepatic damage
is severe or is associated with common bile duct obstruction.
•
Subclinical clotting abnormalities may become clinically apparent after liver biopsy,
surgery, and development of gastroduodenal ulcers.
•
Potential mechanisms for bleeding include primary failure of the hepatocytes to synthesize
clotting factors, vitamin K deficiency, and disseminated intravascular coagulation
(DIC) (see Chapter 23).
Hepatic Encephalopathy
•
HE is a metabolic encephalopathy that occurs secondary to severe liver disease or
portosystemic shunting of blood.
•
Clinical signs include depression, hypersalivation, behavioral changes, altered consciousness,
motor disturbances, seizures, and coma. As with other metabolic encephalopathies,
signs typically wax and wane and are interspersed with normal periods.
•
Ammonia, mercaptans, short-chain fatty acids, gamma-aminobutyric acid (GABA), and
endogenous benzodiazepines are potential encephalopathic toxins that are produced
in the colon by bacterial action on various substrates. Because the liver normally
detoxifies these substances, systemic concentrations are low. With severe liver disease
or portosystemic shunting, these potential toxins reach high concentrations in the
systemic circulation and the central nervous system (CNS), resulting in clinical signs.
•
Exacerbation of encephalopathy occurs after eating a meal high in protein because
protein is a substrate for toxins such as ammonia and mercaptans.
•
HE must be differentiated from other metabolic encephalopathies and primary CNS disorders.
Ascites
•
Ascites is a common feature of severe chronic liver disease. Mechanisms of ascites
and edema formation in liver disease include hypoalbuminemia, portal hypertension,
and sodium and water retention.
•
Rupture of the biliary tract causing bile peritonitis also is associated with abdominal
fluid accumulation.
Signalment and History
•
The signalment often provides important clinical information, because breed predilections
for specific liver diseases have been recognized, and young animals are more likely
to be presented for congenital hepatic disorders such as portosystemic shunt.
•
The history is helpful to characterize the clinical course of liver disease as acute
or chronic. Recent onset of signs in an animal that was previously healthy suggests
acute hepatic failure. However, because of the large functional reserve capacity of
the liver, in occult chronic liver disease the clinical signs may be vague and may
not be recognized by the owner until the final phase of hepatic decompensation.
Key Point
Chronic hepatic disease can be associated with recent onset of clinical signs and
can initially seem to be an acute disease. However, persisting signs of weight loss
and ascites and diagnostic findings of hypoalbuminemia and microhepatica are indicative
of chronic hepatic disease.
•
The history may provide important information regarding the potential for exposure
to known causes of hepatic injury such as drug therapy, surgical and anesthetic procedures,
and toxins or infectious agents.
•
Determine if the animal has a history of intolerance to drugs normally metabolized
by the liver, such as sedatives, tranquilizers, anticonvulsants, and anesthetics.
•
Determine the current vaccination status and exposure potential for infectious agents
known to affect the liver, such as leptospirosis, infectious canine hepatitis, and
feline infectious peritonitis (FIP).
Physical Examination
Skin and Mucous Membranes
•
Evaluate the sclera, oral mucous membranes, and skin for jaundice. Jaundice is not
clinically detectable until serum bilirubin concentrations are >2.5 to 3.0 g/dl. In
cats, subtle jaundice often is best detected on the palatine mucosa.
•
Evaluate the skin and mucous membranes for evidence of bleeding. Pallor may be detected
with blood loss anemia.
Abdominal Palpation
Palpate the abdomen carefully. The normal liver can be difficult to palpate in dogs
and cats, and the edges are normally sharp, not rounded.
•
Hepatomegaly is caused by passive venous congestion, diffuse inflammation, nodular
hyperplasia, cysts, bile engorgement, marked biliary hyperplasia (cats), and infiltration
by fat, glycogen, and neoplastic cells.
•
Pain on palpation of the liver (hepatodynia) usually indicates acute liver disease.
The pain is caused by stretching of the liver capsule and must be differentiated from
pain arising in the pancreas, stomach, or spleen.
•
Moderate to severe abdominal effusion may be detected.
Neurologic Exam
Perform a neurologic examination in animals with a history of neurologic signs. With
HE, the neurologic examination may be normal or suggestive of diffuse cerebral disease
(e.g., depression and dementia, disorientation, pacing, circling, head pressing, hypersalivation,
seizures, or coma).
Rectal Exam
Perform a rectal examination and obtain a fecal sample to evaluate for melena (indicative
of GI bleeding) and acholic feces.
Routine Laboratory Evaluations
Because clinical findings in hepatobiliary disease often are vague, hepatic disease
may not be suspected until biochemical tests identify elevated liver enzyme activity
or other evidence of hepatic dysfunction (e.g., hyperbilirubinemia or hypoalbuminemia).
Liver function studies such as serum bile acid (SBA) concentrations are used to achieve
the following:
•
Identify occult liver disease
•
Assess liver function when there is increased liver enzyme activity but normal serum
bilirubin concentrations
•
Determine whether significant hepatic dysfunction is present to warrant performing
a liver biopsy
•
Monitor response to therapy
Findings consistent with liver disease on routine laboratory tests are described below.
Complete Blood Count
•
Mild to moderate anemia may occur secondary to liver disease because of blood loss
(e.g., gastroduodenal ulceration or coagulopathy) or may be associated with normocytic-normochromic
anemia of chronic disease.
•
Erythrocytic microcytosis is a common finding in dogs and cats with portosystemic
shunts. Decreased serum iron concentration, normal to increased serum ferritin concentration,
and accumulation of stainable iron in the liver suggests that microcytosis is associated
with abnormal iron metabolism (impaired iron transport or sequestration of iron) rather
than absolute iron deficiency. Decreased availability of iron for hemoglobin synthesis
appears to occur despite adequate tissue iron stores.
•
Target cells and poikilocytosis (acanthocytes and elliptocytes) may occur in dogs
and cats with various types of hepatic disease, due to altered red blood cell (RBC)
membranes.
Urinalysis
•
Urine specific gravity may be isosthenuric or hyposthenuric if liver disease is associated
with PU/PD.
•
Bilirubinuria is a sensitive indicator of abnormal bilirubin metabolism, and this
finding precedes hyperbilirubinemia and jaundice. Bilirubinuria imparts a yellow-orange
color to the urine. Bilirubin crystals may form in the presence of bilirubinuria.
•
Trace amounts of bilirubin may be found in concentrated urine of normal dogs (especially
males).
•
Bilirubinuria is always abnormal in cats and suggests underlying hemolytic or hepatobiliary
disease.
•
Urine urobilinogen is a colorless product of enteric bacterial degradation of bilirubin
that is absorbed from the gut. A small portion of urobilinogen escapes the enterohepatic
circulation and is excreted in the urine. The finding of urobilinogenuria indicates
an intact enterohepatic circulation of bilirubin pigments. The absence of urobilinogenuria
in a jaundiced animal suggests common bile duct obstruction. However, this test is
not reliable in a clinical setting because many non-hepatic factors affect urine urobilinogen
concentration, including altered intestinal flora, GI bleeding, intestinal absorption,
renal excretion, urine pH, urine volume, and urine storage.
•
Ammonium biurate crystals are commonly detected in animals with portosystemic shunts.
Key Point
Suspect liver disease in any cat or dog with ammonium biurate crystalluria (except
in dalmatians and bulldogs).
Liver Enzymes
Evaluation of serum liver enzyme activity is used as a screening test to detect liver
disease. Increases in liver enzyme activity are not specific for the underlying hepatic
disorder. However, liver enzymes can be used to categorize the underlying pathophysiologic
mechanism. Increases in liver enzyme activity may occur secondary to hepatocellular
injury and leakage (Fig. 71-1
), or due to increased production stimulated by bile retention (cholestasis) or drug
induction (Fig. 71-2
).
Figure 71-1
With hepatocyte injury, leakage of alanine aminotransferase (ALT) from the cytoplasm
results in increased serum activity. Aspartate aminotransferase (AST) is primarily
associated with mitochondria but is also present in the cytoplasm. Release of AST
from the mitochondria requires a severe insult. Thus, with hepatocyte injury, ALT
is more readily released and its activity level will usually be higher than that of
serum AST.
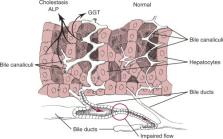
Figure 71-2
Impaired bile flow (cholestasis) causes increased synthesis of alkaline phosphatase
(ALP) and gamma-glutamyltransferase (GGT). ALP is a sensitive indicator of cholestasis
in dogs but is less sensitive in cats (see text). With cholestatic disorders, increased
ALP activity precedes hyperbilirubinemia. ALP and GGT lack specificity in differentiating
between intrahepatic and extrahepatic cholestasis.
Many systemic diseases can secondarily affect the liver (reactive hepatopathy), causing
increased liver enzyme activity, but these are not necessarily associated with clinical
liver disease. For example, feline hyperthyroidism is commonly associated with increased
liver enzyme activity without significant hepatic dysfunction.
Key Point
Liver enzymes do not evaluate liver function. Thus, severe hepatic dysfunction may
coexist with normal liver enzyme activity; conversely, increased liver enzyme activity
may be detected in animals without significant hepatic dysfunction.
Alanine Aminotransferase
Increased alanine aminotransferase (ALT) activity indicates hepatocyte injury with
leakage of enzyme from the cytoplasm of the hepatocyte (see Fig. 71-1). The magnitude
of ALT increase generally correlates with the number of injured hepatocytes.
•
The largest increases in ALT activity occur with hepatocellular necrosis and inflammation
(up to 100 times normal). Increases also occur with increased hepatocyte membrane
permeability, such as that caused by hypoxia. Severe cholestasis can also cause secondary
hepatocyte injury, with increases in ALT up to 20 to 40 times normal. Increased production
of ALT by regenerating hepatocytes may account for persisting increases in enzyme
activity after resolution of the initial injury.
•
Anticonvulsant drug therapy (primidone, phenytoin, and phenobarbital) in dogs can
be associated with mild increases in ALT activity (2 times normal) in the absence
of obvious hepatocellular injury.
•
Corticosteroid therapy or hyperadrenocorticism is associated with mild to moderate
(2-10 times normal) increases in ALT activity.
•
Small amounts of ALT are present in canine skeletal muscle; it has been shown that
severe skeletal muscle degeneration or necrosis (canine muscular dystrophy, necrotizing
myopathy, polymyositis) may be associated with increases in ALT activity of 5 to 25
times normal. When increased ALT activity is caused by muscle injury, creatine kinase
(CK) and AST activity are also markedly increased. Whether ALT is liver specific in
cats remains to be determined.
Aspartate Aminotransferase
Hepatocyte injury is associated with increased AST activity secondary to leakage from
mitochondria and cytoplasm of hepatocytes (see Fig. 71-1).
•
AST is not liver specific in dogs and cats; it is present in significant quantities
in hepatocytes and skeletal muscle tissue.
•
Comparison of activities of ALT, AST, and CK, a muscle enzyme, can indicate whether
AST activity is increased due to hepatic or muscle injury.
Key Point
Increased AST activity associated with hepatic injury generally parallels but is less
than the increase in ALT activity, and CK is normal. Increased AST activity due to
skeletal muscle injury is associated with increased CK activity and normal or mildly
increased ALT activity.
•
In some cats with liver disease, AST may be more sensitive than ALT in detecting hepatic
disease.
Alkaline Phosphatase
Increases in serum alkaline phosphatase (ALP) activity are due to accelerated production
of this enzyme, stimulated by cholestasis or drug induction (see Fig. 71-2). ALP is
a membrane-associated enzyme present in many tissues; however, only liver, bone, and
corticosteroid-induced isoenzymes contribute to serum ALP activity. Serum ALP activity
in normal dogs and cats is usually due to the liver isoenzyme. An increase in this
type of ALP activity indicates intrahepatic or extrahepatic cholestasis.
•
Young growing animals or animals with severe bone disease may have mild increases
in ALP activity due to the bone isoenzyme.
•
Cats generally have smaller increases in serum ALP activity with hepatobiliary disease
than do dogs owing to a limited capacity for ALP production and a shorter serum half-life
(6 hours in cats versus 72 hours in dogs). Therefore, even small increases in serum
ALP activity (2-3 times normal) in cats suggest significant cholestasis.
•
Exogenous or endogenous glucocorticoids are associated with hepatic production of
a novel isoenzyme of ALP, corticosteroid-induced ALP (CIALP), in dogs but not in cats.
The CIALP isoenzyme can be distinguished from the liver isoenzyme (LALP) by levamisole
inhibition using an automated analyzer. Increased CIALP activity is a consistent finding
in dogs with spontaneous hyperadrenocorticism and absence of this isoenzyme is uncommon
in this disorder.
Key Point
Hypercortisolism caused by glucocorticoid therapy or hyperadrenocorticism (Cushing's
disease) is the most common pathologic cause of increased serum ALP activity in dogs;
it is usually attributed to an increase in CIALP.
•
Increase in ALP activity associated with corticosteroid therapy varies considerably
with the individual dog and the drug, dose, and duration of therapy. In the first
7 to 10 days of oral or parenteral glucocorticoid therapy, increases in ALP are primarily
due to LALP activity. By 7 days, CIALP activity begins to increase, and it predominates
in the serum after a month of therapy. Chronic treatment with otic, ophthalmic, and
topical preparations is also capable of inducing ALP activity. Increases in CIALP
activity do not necessarily imply the presence of iatrogenic hyperadrenocorticism,
a suppressed pituitary-adrenal axis, or corticosteroid-induced hepatopathy, nor does
it indicate that corticosteroid therapy must be discontinued.
•
Increased CIALP activity is a sensitive but not a specific test for exposure to excess
glucocorticoids (iatrogenic or endogenous). Increases in CIALP activity may be detected
with diabetes mellitus, anticonvulsant drug therapy, primary hepatic disorders including
neoplasia, hypothyroidism, and chronic illnesses (associated with disease-related
stress and increased endogenous cortisol secretion). In this setting, a mixed pattern
of increased CIALP and LALP activity is seen.
•
Increased CIALP activity associated with exogenous or endogenous glucocorticoids may
be accompanied by mild to moderate (2-10 times normal) increases in ALT activity that
typically are of lesser magnitude than increases in ALP activity.
•
Anticonvulsant drug therapy is associated with enzyme induction of the liver isoenzyme
of ALP in dogs (but not in cats) in the absence of obvious hepatocellular injury.
CIALP activity also may be increased in some dogs. Reported maximal increases for
induced serum ALP activity include those caused by phenobarbital (30 times normal),
primidone (5 times normal), and diphenylhydantoin (3 times normal).
Gamma-Glutamyltransferase
This membrane-associated enzyme is present in many tissues. Increased serum gamma-glutamyltransferase
(GGT) activity usually reflects cholestasis and increased production by hepatocytes
(see Fig. 71-2).
•
Increased GGT activity parallels increased ALP activity in dogs, including increases
associated with excess corticosteroids.
•
Anticonvulsant therapy causes mild (2-3 times normal) increases in serum GGT activity
in dogs.
•
In cats, serum GGT is more sensitive for detecting hepatobiliary disease than ALP.
Serum GGT activity exceeds serum ALP activity in most hepatobiliary diseases; an exception
is hepatic lipidosis, in which GGT activity may be normal or mildly increased despite
a moderate to severe elevation of serum ALP.
Other Biochemical Tests
Numerous biochemical tests can be altered by liver disease, including serum bilirubin,
albumin, globulin, urea nitrogen, glucose, and cholesterol. Many of these parameters
reflect some aspect of liver function; however, they lack sensitivity or specificity
for liver disease.
Bilirubin
Increased serum bilirubin concentration occurs secondary to hemolysis or cholestasis.
Evaluate for underlying hemolytic disorders by performing a complete blood count (CBC)
to detect anemia.
•
Fractionation of the total serum bilirubin into conjugated and unconjugated components
(van den Bergh test) to distinguish the mechanism of hyperbilirubinemia is of little
diagnostic value because there is considerable overlap in hemolytic, hepatocellular,
and extrahepatic biliary disorders.
•
Lipemia falsely elevates serum bilirubin concentration; the absence of concurrent
bilirubinuria suggests pseudohyperbilirubinemia.
Albumin
Albumin is synthesized exclusively by the liver. Because of a large reserve capacity
for albumin production, hypoalbuminemia does not occur until the functional hepatic
mass is reduced 70% to 80%.
Key Point
Hypoalbuminemia associated with hepatic disease implies chronicity because of the
long half-life of albumin.
•
With chronic liver disease, fluid retention and dilution of existing serum albumin
may also contribute to hypoalbuminemia.
•
When the serum albumin is >1.5 g/dl, hypoalbuminemia contributes to the development
of ascites and edema.
•
Hypoalbuminemia is not specific for liver disease, and other causes of hypoalbuminemia
such as urinary and gastrointestinal (GI) loss must be excluded.
•
Inappropriate dietary protein restriction should be avoided since it can worsen hypoalbuminemia.
Globulin
•
Hyperglobulinemia due to increased gamma globulins occurs in some dogs and cats with
chronic liver disease. The most likely mechanism is a systemic response to antigens
that escape from the GI tract because of impaired hepatic mononuclear phagocyte system
function or portosystemic shunting.
•
Significant hypoglobulinemia does not usually occur with liver disease despite the
liver's role in the synthesis of alpha and beta globulins.
Blood Urea Nitrogen
Blood urea nitrogen (BUN) concentration may be decreased secondary to liver disease
because the liver is responsible for converting ammonia to urea. However, many non-hepatic
factors (e.g., PU/PD, fluid diuresis, and low-protein diet) can also decrease BUN
levels.
Glucose
Hypoglycemia may occur secondary to hepatic dysfunction because of impaired hepatic
gluconeogenesis, decreased hepatic glycogen stores, and decreased hepatic insulin
degradation. However, because <30% of liver function is sufficient to maintain euglycemia,
hypoglycemia is an insensitive indicator of hepatic function.
•
Because it indicates severe liver dysfunction, liver-associated hypoglycemia is a
poor prognostic factor, except in dogs and cats with congenital portosystemic shunts.
•
Some hepatic neoplasms such as hepatocellular carcinoma and adenoma, leiomyosarcoma,
and hemangiosarcoma have been associated with profound hypoglycemia.
•
Also consider non-hepatic causes of hypoglycemia such as sepsis, hypoadrenocorticism,
and insulinoma (see Chapters 33 and 35).
Cholesterol
•
Hypercholesterolemia occurs with acute cholestatic disorders because of increased
synthesis of cholesterol and decreased incorporation of cholesterol into bile acids;
however, there are many non-hepatic causes of hypercholesterolemia.
•
Although cholesterol is synthesized in the liver, hypocholesterolemia secondary to
liver disease is uncommon; it has been noted with congenital portosystemic shunts
and phenobarbital-induced hepatic disease.
Electrolytes
Serum electrolyte changes secondary to liver disease are variable.
•
In acute liver failure, serum electrolyte concentrations are usually normal.
•
With chronic liver disease, total body potassium depletion and sodium and water retention
are common, and the serum sodium concentration is usually normal or decreased.
Liver Function Tests
Liver function tests can document clinically significant hepatic dysfunction when
liver disease is suspected, based on historical, clinical, laboratory, and radiographic
findings. SBA determinations have largely replaced the use of organic anion dyes such
as sulfobromophthalein (Bromsulphalein) and indocyanine green (ICG). Blood ammonia
concentration and ammonia tolerance tests can specifically evaluate the portal circulation
(for portosystemic shunts) and detect HE.
Key Point
The test of choice for clinical evaluation of liver function is the combined fasting
and 2-hour postprandial SBA test.
Serum and Urine Bile Acids
The normal physiology of bile acid metabolism is shown in Figure 71-3A
In health, bile acids are confined to the enterohepatic circulation, and systemic
concentrations are low. SBA concentrations increase in the systemic circulation with
all types of liver disease (Fig. 71-3B). Because the liver has a large reserve capacity
for synthesis of bile acids, even severe hepatic dysfunction does not cause decreased
SBA concentrations.
Figure 71-3
A, Bile acids are synthesized in the liver, secreted into the biliary system, and
stored in the gallbladder during fasting. With ingestion of a meal, cholecystokinin
release stimulates gallbladder contraction and entry of bile acids into the intestinal
tract. Bile acids are efficiently reabsorbed in the distal ileum and carried in the
portal blood back to the liver, thus completing the enterohepatic circulation. In
the healthy animal, the liver removes 90% to 95% of bile acids from the portal circulation
during the first pass of the enterohepatic circulation. This allows only small amounts
of bile acids to escape to the systemic circulation. Normal serum concentrations are
therefore low (fasting < 15μmol/L, postprandial < 25μmol/L). B, Hepatocellular dysfunction
or cholestasis interferes with hepatic uptake, storage, and secretion of bile acids.
Thus, impaired extraction of bile acids from the portal blood results in increased
serum bile acid concentrations. With portosystemic shunting, bile acids in the portal
blood are diverted directly into the systemic circulation.
Fasting Serum Bile Acid Concentration
A fasting serum bile acid (FSBA) concentration obtained after a 12-hour fast is a
sensitive, specific measure of hepatobiliary function in dogs and cats. Normal FSBA
values in dogs and cats are <20μmol/L. When concentrations exceed 30μmol/L, a liver
biopsy may be warranted to evaluate the underlying liver disease.
•
Increased concentrations occur with hepatocellular and cholestatic disorders that
interfere with hepatic uptake or secretion of bile acids and with portosystemic shunting,
in which bile acids are diverted directly into the systemic circulation (see Fig.
71-3B).
•
Serial evaluation of SBA concentration to monitor liver function may not have merit
unless the SBA concentration returns to normal. This is because there are wide fluctuations
in SBA concentration in a given patient with liver disease within a 24-hour period
(although all values are abnormal).
•
Dogs and cats receiving the oral synthetic bile acid, ursodiol, may have an increase
in SBA concentration because of the absorption of the drug and reflection of its presence
in the serum.
•
Interference in bile acid measurement can occur with hemolysis or lipemia.
•
In dogs with unexplained increases in SBA, consider the possibility of small intestinal
bacterial overgrowth causing increases in unconjugated bile acids.
•
Healthy Maltese dogs were reported to have significantly higher SBA (mean 70μmol/L
α50; range 1–362μmol/L) as measured by the enzymatic spectrophotometric method than
as measured by high-performance liquid chromatography, suggesting the presence of
additional reacting substances or unusual bile acids in their serum. However, a methodological
problem related to lipemia or hemolysis cannot be excluded. It is also possible that
these dogs had underlying hepatic microvascular dysplasia.
•
When hepatic disease causes hyperbilirubinemia, measurement of SBA concentrations
does not provide any additional diagnostic information. SBA concentrations are most
helpful diagnostically in dogs and cats with anicteric liver disease.
Postprandial Serum Bile Acid Concentration
Postprandial serum bile acid (PPSBA) concentration is an endogenous challenge test
of liver function. Whether PPSBA concentration is a more useful diagnostic test than
FSBA concentration remains unclear. In dogs, similar information is provided by either
test in most hepatobiliary disorders. Notable exceptions include dogs with portosystemic
shunts or cirrhosis, because with these disorders, FSBA can be in the normal range.
In cats, the diagnostic efficacy of PPSBA exceeds that of FSBA for all hepatic disorders,
including portosystemic shunts. For best diagnostic utility, paired FSBA and 2-hour
PPSBA is recommended. To perform the PPSBA concentration test, take the following
steps:
•
Obtain a serum sample for FSBA concentration, and then feed at least 2 teaspoons of
food to small dogs and cats (<5 kg) and at least 2 tablespoons to larger patients.
•
To ensure gallbladder contraction, feed a diet high in fat (e.g., Hill's Pet Prescription
Diet p/d and Hill's Pet Prescription Diet c/d for cats). For encephalopathic animals
in which a high-protein diet is contraindicated, substitute a protein-restricted diet
and add a few milliliters of corn oil per feeding.
•
Obtain a second serum sample 2 hours after feeding.
•
Results: In normal dogs and cats, PPSBA concentrations are <25μmol/L and peak 2 hours
after a meal. A liver biopsy may be indicated when concentrations are >30μmol/L.
•
On occasion, FSBA is higher than PPSBA. This probably occurs because of sporadic gallbladder
contraction during fasting, which releases bile into the intestinal tract, resulting
in increased SBAs.
Urine Bile Acids
Recently, urine bile acids (UBAs) have been investigated as a diagnostic tool in dogs
and cats. Normally only small amounts of bile acids are present in the urine. Liver
disease and increased SBA result in increased excretion of bile acids in the urine.
Potential advantages of UBA over a random FSBA are that UBA may reflect an average
value over time, ease of sample collection, and lack of interference from oral ursodiol
administration.
•
UBAs are normalized with urine creatinine (Cr), and values are expressed as UBA/Cr
(μmol/mg) × 100. Values greater than 7.3 are abnormal in dogs. Values greater than
4.4 are abnormal in cats.
•
Timing of urine collection to the 4- to 8-hour post-prandial period may improve diagnostic
performance, particularly in dogs with congenital portosystemic shunting where sensitivity
of UBA (taken 4 to 8 hours after eating) was 100% compared with 84% for FSBA and 98%
for PPSBA.
•
The role of UBA in the evaluation of hepatobiliary function awaits further clinical
evaluation.
Blood Ammonia Concentration
Ammonia is metabolized by the liver, and normal plasma concentrations are low. Measurement
of ammonia is technically difficult, and appropriate sample handling requires heparinized
blood samples to be stored immediately on ice, cold-centrifuged, and assayed as soon
as possible.
•
Measurement of blood ammonia concentration primarily is indicated to document HE.
However, normal values do not exclude this diagnosis, because other toxins can contribute
to the encephalopathy.
•
Portosystemic shunting (congenital or acquired) is the most common mechanism of hyperammonemia,
but severe, diffuse hepatic disease (especially acute hepatic necrosis) also increases
blood ammonia concentrations.
•
Blood ammonia values have poor sensitivity in detecting other types of hepatobiliary
disease without portosystemic shunting.
•
Blood ammonia concentration is not a suitable screening test for congenital portosystemic
shunt in young Irish wolfhounds since a transient metabolic hyperammonemia unassociated
with liver disease occurs in this breed.
•
Congenital urea cycle enzyme deficiency is a rare cause of hyperammonemia.
Ammonia Tolerance Test
This is a more sensitive test than blood ammonia concentration for documenting portosystemic
shunting. However, the ammonia tolerance test (ATT) is contraindicated if resting
ammonia levels are already increased, because no further diagnostic information will
be obtained and performing an ATT can cause signs of HE. Note: The ATT is not recommended
for use in cats.
•
The test is performed in a fasted dog by giving 100 mg/kg of ammonium chloride (do
not exceed a total dose of 3 g), either as a dilute solution by stomach tube or as
a powder in a gelatin capsule.
•
Take a heparinized blood sample before and 30 minutes (stomach tube method) or 45
minutes (capsule method) after administering the ammonium chloride. Vomiting may occur
but does not invalidate the test.
•
In normal dogs, there is no increase in blood ammonia concentration or a mild increase
(<2 times greater than baseline). In dogs with portosystemic shunting, results are
consistently abnormal (up to 10 times baseline values).
•
The ATT and paired FSBA and PPSBA tests have equal sensitivity in detecting portosystemic
shunting. Consequently, the SBA testing has largely replaced blood ammonia and ATT.
Paired FSBA and PPSBA provide a better screening test because it detects a broader
range of hepatobiliary disorders, and bile acids are stable, permitting routine laboratory
analysis.
Protein C
Protein C, an anticoagulant protein synthesized in the liver, has been investigated
as a clinical marker of liver disease. Preliminary results show decreased protein
C concentrations in 100% of dogs with liver failure, 98% of dogs with portosystemic
shunt, and 30% of dogs with hepatic microvascular dysplasia. The role of protein C
in the detection of liver disease awaits further clinical studies.
Parameters of Hemostasis
The liver plays a central role in the coagulation and fibrinolytic systems. The liver
is responsible for synthesis of all coagulation factors except factor 8, von Willebrand
factor. Fibrinogen, antithrombin, and protein C are all synthesized in the liver and
can be decreased with hepatic dysfunction. Activated coagulation factors and fibrinolytic
enzymes are also cleared by the liver.
•
Mechanisms of excessive bleeding associated with hepatobiliary disease include primary
failure of hepatocytes to synthesize clotting factors, DIC, and vitamin K deficiency.
Vitamin K deficiency in hepatobiliary disease is usually caused by malabsorption of
vitamin K secondary to complete bile duct obstruction. However, a vitamin K–responsive
coagulopathy may sometimes be detected in dogs and cats with severe hepatic insufficiency,
possibly due to marked intrahepatic cholestasis causing vitamin K malabsorption or
the inability of the liver to reactivate vitamin K from its inactive (epoxide) form.
•
Clinical evidence of bleeding secondary to hepatobiliary disease is uncommon; however,
the frequency of abnormal coagulation tests is much higher.
Coagulation Tests
•
Measure prothrombin time (PT) to evaluate the extrinsic coagulation system and activated
partial thromboplastin time (APTT) to evaluate the intrinsic coagulation system. These
tests can be abnormal in the presence of liver disease. Activated coagulation time
also can be used as a rapid screening test for abnormalities of the intrinsic coagulation
system. For further discussion of these tests, see Chapter 23.
•
The PIVKA (proteins induced by vitamin K absence) clotting time is a more sensitive
test than PT or APTT to detect bleeding tendencies in dogs and cats with liver disease.
Normalization of PIVKA clotting time may occur after treatment with vitamin K1.
•
The combination of prolonged PT and APTT, low plasma fibrinogen, increased fibrin
degradation products, fragmented RBCs, and thrombocytopenia suggests DIC (see Chapter
23).
Thrombocytopenia and Platelet Dysfunction
•
Thrombocytopenia may occur secondary to splenic sequestration of platelets associated
with portal hypertension or consumption of platelets from DIC.
•
Platelet function defects also have been documented in dogs with liver disease, which
may account for clinical bleeding tendencies in the presence of normal coagulation
tests and platelet numbers. Use mucosal bleeding time to detect platelet function
abnormalities.
Blood Gas Analysis
Various acid-base imbalances may occur secondary to liver disease, including respiratory
alkalosis, metabolic alkalosis, metabolic acidosis, and mixed acid-base disturbances.
Abdominal Fluid Analysis
•
Ascitic fluid that accumulates secondary to liver disease and hypoalbuminemia is usually
a transudate. With hepatic venous congestion from vena caval obstruction or cardiac
causes, the fluid typically is a modified transudate with protein concentration >
2.5 g/dl.
•
Rupture of the biliary tract is associated with bile peritonitis. Grossly, the abdominal
fluid appears yellow or green. Chemical tests for bilirubin are positive, and concentrations
of bilirubin are higher in abdominal fluid than in serum. Cytologic examination reveals
a mixed inflammatory infiltrate and bile-laden macrophages. Bacteria may be seen if
bile peritonitis is complicated by sepsis.
Diagnostic Imaging
Survey Radiography
Abdominal radiographs are useful to evaluate for the following:
•
Changes in liver size (hepatomegaly, microhepatica)
•
Altered tissue characteristics such as mineralized hepatic densities (choleliths)
and radiolucencies (abscesses)
•
Presence of abdominal effusion
If hepatic neoplasia is suspected, take thoracic films to evaluate for pulmonary metastases.
Ultrasonography
Ultrasonography can be used to image the liver non-invasively, especially when abdominal
effusion precludes survey radiographic evaluation. A normal ultrasonographic appearance
of the liver does not eliminate the possibility of significant hepatic pathology;
however, ultrasonography is diagnostically useful to achieve the following:
•
Detect focal parenchymal abnormalities such as masses, abscesses, cysts, and regenerative
nodules. The ultrasonographic appearance of these focal lesions often is similar,
and biopsy is required for differentiation.
•
Document that a palpable mass is associated with the liver.
•
Investigate disorders of the biliary tract and gallbladder, such as biliary obstruction,
cholelithiasis, or gallbladder mucocele.
•
Detect vascular lesions such as portosystemic shunts, hepatic arteriovenous fistulas,
and hepatic venous congestion.
•
Obtain percutaneous liver biopsies (see below).
•
Identify abnormalities in other abdominal organs that may be a cause or effect of
liver disease (e.g., pancreas, spleen, kidneys, bladder, GI tract, adrenal glands,
and lymph nodes). For example, urate or uric acid urolithiasis may be detected in
animals with portosystemic shunts, the primary tumor may be identified in animals
with hepatic metastases, and identification of adrenal gland enlargement may aid the
diagnosis of steroid hepatopathy.
Angiography
Angiography is useful to diagnose vascular disorders involving the liver, such as
congenital and acquired portosystemic shunts, hepatic arteriovenous fistulas, and
vena caval obstruction causing hepatic venous obstruction (see under “Congenital Portosystemic
Shunt”).
Liver Cytology
Fine-needle aspiration (FNA) of the liver for cytology is commonly performed because
it is easy and safe, does not require sedation or anesthesia, and provides rapid preliminary
information. However, the diagnostic accuracy of cytology versus histopathology of
the liver is controversial. Studies suggest a lack of correlation exists as much as
50% of the time. Cytology of impression smears of liver biopsy tissue correlates better
than samples obtained by fine-needle aspirate.
•
Cytology of the liver is most useful if the pathologic process is diffuse and architectural
relationships (which can be obtained only by histopathology) are not essential to
the diagnosis. Examples include vacuolar hepatopathies such as feline hepatic lipidosis,
diffuse hepatic neoplasia, and liver disease associated with infectious agents such
as histoplasmosis.
•
In cats, primary liver diseases such as lymphoma and cholangitis may be accompanied
by hepatic vacuolar changes. These cats may be misdiagnosed as idiopathic hepatic
lipidosis if cytology only reflects the vacuolated hepatocytes.
•
For focal lesions, accuracy is improved when the fine-needle aspirate is guided by
ultrasound.
•
Poor correlation of cytology with histopathology occurs in primary inflammatory liver
diseases, although it may be better for detection of suppurative inflammation than
for lymphocytic-plasmacytic inflammation.
Liver Biopsy
Liver biopsy often is required to definitively characterize the nature and severity
of hepatic disease, to differentiate acute from chronic disorders, and to assess response
to therapy. Selection of the best procedure for obtaining a liver biopsy depends on
numerous factors, including liver size, presence of coagulopathy, diffuse versus focal
hepatic lesions, presence of biliary tract obstruction, presence of other intra-abdominal
abnormalities, likelihood of surgical resection of a mass, tolerance of general anesthesia,
available equipment, and expertise of the clinician.
Key Point
Perform a hemostasis screen prior to liver biopsy to detect coagulopathy. After the
biopsy is performed, monitor for bleeding from the biopsy site.
Biopsy Methods
Ultrasound-Guided Needle Biopsy
This technique is the most common percutaneous method used for liver biopsy. However,
it is dependent on the availability of equipment and clinician expertise.
•
With ultrasound-guided biopsy, it is possible to obtain tissue from focal lesions
(whether superficial or deep within the hepatic parenchyma), avoid structures adjacent
to the liver, and monitor post-biopsy bleeding.
•
Ultrasound-guided biopsy may be difficult if the liver is small or the ultrasonographer
lacks experience.
•
Because needle biopsy specimens are smaller than wedge biopsies, they may not be representative
of underlying liver pathology. In one study, the morphologic diagnosis made by needle
biopsy correlated with the definitive diagnosis obtained by wedge biopsy in only 48%
of dogs and cats.
Laparoscopy
Laparoscopy provides direct visualization of the liver and adjacent structures such
as the pancreas and extrahepatic biliary tract. Biopsies also are obtained under direct
visualization.
•
Laparoscopy is a useful alternative to ultrasound-guided needle biopsy when the liver
is small.
•
It is preferable to ultrasound-guided biopsy when excess bleeding is anticipated and
to laparotomy when delayed wound healing (hypoalbuminemia) is anticipated.
•
Laparoscopy requires heavy sedation or anesthesia and is subject to equipment availability
and clinician expertise.
Laparotomy
Laparotomy is indicated for liver biopsy when a surgically correctable disease is
suspected, such as extrahepatic biliary tract obstruction or a single, large hepatic
mass (see Chapter 72 for a description of the procedure for surgical biopsy).
•
Laparotomy makes it possible to obtain large samples of liver tissue and monitor for
post-biopsy bleeding.
•
Disadvantages include the need for general anesthesia, the relatively high risk of
complications, and the risk of delayed wound healing in hypoalbuminemic patients.
Biopsy Analysis
•
To prepare biopsy tissue for histopathology, place samples in 10% buffered formalin
and allow them to fix for 24 hours. The volume of fixative should be 20 times the
volume of biopsy tissue.
•
To prepare needle biopsy samples, gently remove liver tissue from the biopsy needle
and place it on tissue paper; fold the paper and place it in a formalin jar.
•
Perform routine light microscopy on liver tissue stained with hematoxylin and eosin
(H&E).
•
Additional stains may be requested, including trichrome for fibrous connective tissue,
periodic acid-Schiff (PAS) for glycogen, rhodanine or rubeanic acid for copper, Prussian
blue for iron, Congo red for amyloid, oil red O for fat, and silver or acid-fast stains
for infectious organisms.
•
Submit fresh liver tissue for bacterial and fungal culture as indicated.
•
Perform quantitative copper analysis on fresh hepatic tissue.
•
Place samples for electron microscopy in chilled, buffered 2.5% glutaraldehyde.
PRINCIPLES OF TREATMENT FOR LIVER DISEASE
Objectives
•
Whenever possible, identify and eliminate the inciting or predisposing causes of liver
disease. Identification of the underlying cause of hepatobiliary disease can provide
insight into specific therapy, the likelihood and nature of potential complications,
and the prognosis for recovery. (Therapy for individual hepatobiliary disorders is
discussed later under specific diseases.)
•
Prevent or manage complications of liver failure, including HE, ascites, GI ulceration,
coagulopathy, infection, and endotoxemia.
•
In patients in which hepatic regeneration and recovery are possible, supportive care
allows time for this to occur. In other cases, clinical manifestations of hepatic
failure may be minimized for variable periods.
Consider Drug Metabolism
The liver is a major site of drug metabolism, and liver disease may alter drug metabolism.
In many cases, hepatic disease is associated with decreased hepatic clearance of a
drug, with subsequent potential toxicity.
Key Point
Prior to administering any drug to a patient with hepatic disease, consider whether
the drug is metabolized or excreted by the liver, is potentially hepatotoxic, or may
exacerbate signs of liver failure.
Avoid drugs that fit the following descriptions:
•
Are known to depend primarily on the liver for inactivation or excretion
•
Are potential hepatotoxins, such as phenobarbital
•
May worsen signs of hepatic failure, such as methionine-containing products, tranquilizers,
sedatives, and diuretics (may exacerbate HE) and nonsteroidal anti-inflammatory drugs
(NSAIDs) and corticosteroids (may cause GI bleeding and exacerbate HE)
Supportive Therapy for Liver Disease
Supportive measures for patients with liver disease are summarized in Table 71-1
.
Table 71-1
GENERAL THERAPY OF HEPATOBILIARY DISEASE
Therapeutic Goals
Therapeutic Regimen
Fluid Therapy
Maintain hydration
Use a balanced polyionic solution such as lactated Ringer's solution or Plasma-Lyte
148 IV.a Use 0.45% NaCl in patients with ascites or edema.
Prevent hypokalemia
Add 20–30 mEq KCl to each liter of maintenance fluid. Monitor serum potassium daily
and adjust as necessary.
Maintain acid-base balance
Avoid alkalosis in HE by using 0.45% or 0.90% saline for IV fluid therapy. Give NaHCO3
or acetate-containing fluids (Plasma-Lyte) rather than lactated fluids for treatment
of severe metabolic acidosis. Avoid lactate-containing fluids in cats with severe
hepatic lipidosis.
Prevent or control hypoglycemia
To treat hypoglycemia, give 50% dextrose (0.5–1 ml/kg) IV to effect. To maintain normoglycemia,
add dextrose to fluids to achieve a 2.5–5.0% solution.
Nutritional Support
Maintain caloric intake
Provide 40–60kcal/kg/day of good-quality diet.
Provide adequate vitamins and minerals
Add B vitamins to fluids of anorexic cats. For long-term therapy, give an oral vitamin-mineral
(especially B vitamin) supplement.
Give vitamin K1 (0.5–1.5 mg/kg IM or SC q12h for three treatments, then weekly as
needed) in biliary obstruction or severe cholestatic liver disease (dogs and cats).
Modify diet to control complications
See specific complications (e.g., HE and ascites).
Control HE
Modify diet
Give NPO in initial stages of HE. For long-term management, provide a reduced-protein
(dairy or vegetable source protein preferred; avoid red meat protein), easily digested,
high-carbohydrate diet. Recommend moderate protein restriction of 15–20% dry matter
(dogs) or 30–35% dry matter (cats). Increase dietary soluble fiber (psyllium 1–3tsp/day).
Prevent formation and absorption of enteric toxins
In hepatic coma, give a warm-water cleansing enema initially (10–20 ml/kg) until fluid
is clear, followed by retention enema (5–10 ml/kg q8–12h) containing lactulose (30%
lactulose with 70% water) and neomycin solution (22 mg/kg) and held for 20–30 minutes
or povidone iodine solution (diluted 1:10 with water, 50–200 ml total) and flushed
out within 10 minutes. For follow-up oral therapy, give neomycinb (22 mg/kg q8–12h
PO), metronidazole (7.5 mg/kg q12h PO), or amoxicillin (22 mg/kg q12h PO) combined
with lactulosec (0.25–0.5 ml/kg q8–12h PO) or lactitol (0.5–0.75 g/kg q12h PO) to
achieve two or three soft stools per day; if diarrhea occurs, reduce dose.
Control gastrointestinal hemorrhage
Correct coagulopathy. Treat Gl parasites and treat gastric ulcer (famotidine or nizatidine
or omeprazoled combined with sucralfatee). Avoid drugs that exacerbate Gl hemorrhage
(e.g., aspirin and other NSAIDs or glucocorticoids).
Correct metabolic imbalances (e.g., dehydration, azotemia, hypokalemia, alkalosis,
and hypoglycemia)
See fluid therapy above.
Avoid drugs or therapies that exacerbate HE
When possible, avoid sedatives, tranquilizers, anticonvulsants, analgesics, anesthetics,
methionine-containing products, diuretics, or stored blood transfusion.
Control seizures
For refractory seizures in dogs, use loading dosages of sodium bromide IV (3% NaBr
600–800 mg/kg over 24 hours) or potassium bromide orally (KBr 100–200 mg/kg q6h for
24 hours), followed by KBr at a dosage of 15–30 mg/kg q12h POf or IV phenobarbitalg
at reduced doses in dogs and cats (monitor serum concentrations to adjust the dose).
Avoid benzodiazepines. For status epilepticus, consider general anesthesia with propofol
to control seizures. Intubate and use mechanical respirator to maintain pO2 and pCO2.
Give mannitol (0.5–1 g/kg by IV bolus over 20 minutes) for suspected cerebral edema.
For chronic, stable seizure management or long-term therapy in dogs, give potassium
bromide (15–30 mg/kg q12h PO in food). For maintenance therapy in cats, consider topiramate
(3.125 mg q12h PO initially, then 6.25 mg q12h PO). For short-term perioperative anticonvulsant
therapy for congenital PSS, consider felbamate (15–20 mg/kg q8h PO), levetiracetam
(20–30 mg/kg q12h), or topiramateh (5–10 mg/kg q12h PO) in dogs or topiramate (3.125
mg q12h PO initially, then 6.25 mg q12h PO) in cats.
Control infection
Give systemic antibiotics (see below).
Control ascites and edema
Give a low-sodium diet; combine furosemide (1–2 mg/kg q12h PO)i and spironolactone
(1–2 mg/kg q12h PO)g, or spironolactone and hydrochlorothiazide (Aldactazide,j 2 mg/kg
q12h PO). Use paracentesis for relief of dyspnea or extreme abdominal distention,
synthetic colloids such as hetastarch (10–20 ml/kg/day IV in dogs and 10–15 ml/kg/day
IV in cats) or human albumin 25% (2 ml/kg in dogs), or plasma transfusion for albumin
replacement (25–45 ml/kg).
Control coagulopathy and anemia
Give vitamin K1 (0.5–1.5 mg/kg q12h, IM or SC for three treatments, then weekly as
needed for dogs and cats); fresh plasma or fresh-frozen plasma (10 ml/kg); or a fresh
blood transfusion (10–15 ml/kg). For DIC, give heparin (75–100 IU/kg q8–12h SC). For
dogs with vWD and liver failure, give DDAVP once (1 mg/kg diluted in 10–20 ml of saline
and given IV slowly over 10 minutes or undiluted SC) (effective for 4–6 hours).
Control gastrointestinal ulceration
Give famotidine (0.5–1.0 mg/kg q12–24h PO or IV), nizatidine (2.5–5.0 mg/kg q24h PO),
or omeprazoled (0.5–1.0 mg/kg q24h PO) and sucralfatee (1-g tablet/25 kg q8h PO).
Control infection and endotoxemia
Give systemic antibiotics (e.g., amoxicillin, ampicillin, cephalosporins, aminoglycosides,
and metronidazolek).
DDAVP, desmopressin acetate; DIC, disseminated intravascular coagulation; GI, gastrointestinal;
HE, hepatic encephalopathy; NPO, nothing per os; NSAIDs, nonsteroidal anti-inflammatory
drugs; vWD, von Willebrand disease.
a
May be given SC if animal is mildly dehydrated and is not vomiting.
b
Neomycin has been associated with rare ototoxicity and nephrotoxicity; its use should
be restricted to acute management of HE.
c
Crystalline lactulose (powder) is also available commercially (Kristalose, Bertek
Pharmaceuticals) as 10- or 20-g packets (syrup concentration is 10 g/15 ml).
d
Partially metabolized by the liver; use a reduced dose in animals with liver failure.
Inhibits hepatic P-450 enzymes.
e
Beware of drug-associated constipation, which may worsen HE.
f
Avoid bromide in cats due to cough and asthma-like side effects.
g
Can be used in dogs and cats with congenital PSS, but avoid in animals with acquired
liver disease.
h
Start at the low end of the dose range due to impaired hepatic metabolism.
i
Dose may be doubled if there is no effect in 4–7 days.
j
GD Searle & Co., Chicago, IL.
k
Partially metabolized by liver. Use a reduced dosage (7.5 mg/kg q12h PO) in animals
with liver failure.
Adapted from Johnson SE: Liver and biliary tract. In Anderson NV (ed): Veterinary
Gastroenterology, 2nd ed. Philadelphia: Lea & Febiger, 1992, p 504.
© 2006 Lea & Febiger
2006
Since January 2020 Elsevier has created a COVID-19 resource centre with free information
in English and Mandarin on the novel coronavirus COVID-19. The COVID-19 resource centre
is hosted on Elsevier Connect, the company's public news and information website.
Elsevier hereby grants permission to make all its COVID-19-related research that is
available on the COVID-19 resource centre - including this research content - immediately
available in PubMed Central and other publicly funded repositories, such as the WHO
COVID database with rights for unrestricted research re-use and analyses in any form
or by any means with acknowledgement of the original source. These permissions are
granted for free by Elsevier for as long as the COVID-19 resource centre remains active.
Restore Fluid, Electrolyte, and Acid-Base Balance
•
Maintain normal fluid balance to support hepatic blood flow and microcirculation and
prevent complications such as HE, DIC, shock, and renal failure. The composition of
the fluid to be given is influenced by the patient's electrolyte and acid-base status
and the presence or potential for hypoglycemia (see Table 71-1 for guidelines; also
see Chapter 5).
•
Avoid alkalinizing agents (e.g., lactate in lactated Ringer's solution and sodium
bicarbonate) when HE is present or impending, because alkalosis augments the entry
of ammonia into the CNS and can exacerbate signs of HE.
Give Nutritional Support
Nutritional support is important for promoting hepatic regeneration and maintenance
of body weight.
•
Modify the diet as needed to control complications of hepatic disease, such as HE,
hypoproteinemia, and ascites (see Table 71-1).
•
Supply the bulk of the calories by carbohydrates, which provide an easily assimilated
source of non-protein calories.
•
Avoid high-protein diets that may exacerbate signs of HE. Indiscriminate protein restriction
is discouraged, however, because adequate protein intake is important for normal hepatic
regeneration and to counteract hypoproteinemia.
•
When voluntary food intake is lacking, provide other methods of nutritional support,
such as feeding through a gastrostomy tube (see Chapter 3).
Control Complications
Hepatic Encephalopathy
Goals for treatment of HE are summarized in Table 71-1. Restrict dietary protein intake.
Increase dietary soluble fiber (see Table 71-1). Prevent formation and absorption
of enteric toxins.
•
Give antibiotics (e.g., neomycin, amoxicillin, or metronidazole) to alter the urease-producing
bacterial population in the colon, thus decreasing conversion of urea to ammonia (see
Table 71-1).
•
Lactulose, a synthetic disaccharide, often is effective in controlling signs of HE
and decreasing arterial blood ammonia concentrations through its actions as a cathartic
and colonic acidifier (see Table 71-1). It usually is given in combination with antibiotics.
Key Point
Detect and control GI hemorrhage, which could provide enteric bacteria with a source
of protein for toxin production. Give fresh rather than stored blood if a transfusion
is required, because stored blood contains substantial amounts of ammonia.
Ascites and Edema
Ascites in liver disease usually is associated with hypoalbuminemia, portal hypertension,
and renal retention of sodium and water.
•
For treatment of ascites, restrict dietary sodium and use diuretics to promote urinary
sodium and water excretion (see Table 71-1).
•
For temporary support of plasma colloid osmotic pressure in hypoproteinemic animals,
consider plasma transfusion to supply albumin or colloid administration such as hetastarch
(see Table 71-1 and Chapter 5).
•
Avoid using abdominocentesis to treat ascites except when required for relieving respiratory
distress.
Coagulopathy and Anemia
Hemostatic defects associated with hepatobiliary disease can be attributed to primary
failure of hepatocytes to synthesize clotting factors, vitamin K deficiency, or DIC.
•
Parenteral administration of vitamin K1 corrects coagulopathy caused by vitamin K
deficiency within 24 to 72 hours, but no response is seen when bleeding is caused
by hepatocyte failure or DIC (see Table 71-1).
•
For treatment of DIC and other coagulopathies, see Chapter 23.
Gastrointestinal Ulceration
Dogs and cats with hepatobiliary disease are at increased risk for GI ulceration.
Possible mechanisms include gastric acid hypersecretion, impaired gastric mucosal
blood flow secondary to portal hypertension, and decreased gastric epithelial cell
turnover.
•
GI bleeding is deleterious in patients with HE because blood is a substrate for ammonia
production.
•
Manage GI ulceration with an H2 blocker for control of acid secretion and with sucralfate
for mucosal cytoprotection, as described in Chapter 67. Famotidine and nizatidine
are preferable to cimetidine and ranitidine as H2 blockers in animals with liver disease,
because they do not inhibit hepatic microsomal enzymes.
Key Point
The proton pump inhibitor, omeprazole, inhibits hepatic microsomal enzymes and undergoes
hepatic metabolism, making it less predictable for use in animals with liver disease.
Infection and Endotoxemia
An increased incidence of infection may be seen in animals with hepatic disease as
enteric bacteria and endotoxins gain access to the systemic circulation as a result
of impaired hepatic mononuclear phagocyte system function or portosystemic shunting.
Septicemia and endotoxemia may, in turn, perpetuate liver injury.
•
Give systemic antibiotics to control extrahepatic infections or sepsis. Penicillins,
cephalosporins, or aminoglycosides are good choices because they are eliminated primarily
by renal mechanisms.
Renal Failure
Renal dysfunction and azotemia may complicate liver disease, especially chronic liver
dysfunction, and can be pre-renal, primary renal, or both.
•
Pre-renal mechanisms that decrease effective circulating volume and renal perfusion
include dehydration (including that induced by diuretics), hypoalbuminemia, ascites,
and overzealous abdominal paracentesis. Appropriate fluid therapy is essential to
avoid pre-renal azotemia.
•
Primary renal failure may occur with pre-existing renal disease or may result from
infectious or toxic agents (e.g., leptospirosis) that affect the liver and kidneys
concurrently or secondary to advanced liver disease.
Hepatoprotectants
Consider ancillary treatment with the hepatoprotectants listed in Table 71-2
. These do not appear to be toxic when used as described.
Table 71-2
HEPATOPROTECTANTS
Product
Preparation
Dosage
Mechanism of Action
Comments
S-adenosylmethionine (SAMe)
Denosyl SD4 (Nutramax Labs): 90 and 225 mg tab
20 mg/kg PO q24h (D&C)
Intermediary metabolite: indirect glutathione precursor (antioxidant), choleretic
(cats), detoxification. Supports membrane function.
Don't break enteric-coated tablets. Use only foil-wrapped products. Food decreases
absorption. No side effects are noted. Can be used in acetaminophen toxicity. Expensive.
Acetylcysteine
10% or 20% solution
Dilute at least 1:4 with saline. Give 140 mg/kg IV through 0.25-mm filter over 20–30
min, then 70 mg/kg PO or IV q6h for seven treatments (D&C)
Glutathione precursor.
Antidote for acetaminophen toxicity. May have protective effects for other drug-induced
(carprofen, potentiated sulfas, methimazole, diazepam) or toxin-induced liver injury.
Safe. May cause nausea and vomiting when given orally.
Milk thistle (Silybin)
Marin (Nutramax Labs: silybin, vitamin E, zinc)* 9-, 24-, or 70-mg tab; Sil-phos (Indea
Labs)
6–100 mg total dose PO q24h per package insert (D); 9–18 mg total dose PO q24h (C)
Antioxidant, anti-inflammatory, antifibrotic. Protects against Amanita mushroom toxicity
(experimental) in dogs.
Silybin has low bioavailability; improved when complexed with phosphatidylcholine
(Marin, Sil-phos). Other products have variable potency and absorption.
Vitamin E (alpha-tocopherol)
Many available
50–400 IU PO q24h (D&C)
Membrane-associated antioxidant. Protects liver against oxidative injury.
Natural Vitamin E (d-a tocopherol) has greater bioavailability than synthetic (d,l)
form. In severe cholestasis, use water-soluble form.
Ursodiol
Actigall (Ciba): 300-mg caps
15 mg/kg PO q24h (D&C)
Hydrophilic bile acid. Shifts bile acid pool to less toxic hydrophilic bile acids.
Choleretic in dogs (cats unknown). Protects hepatocyte membranes. Modulates immune
response.
Used in cholestatic disorders; contraindicated in biliary obstruction. Side effects
are rare (vomiting). Expensive.
L-carnitine
Many available
250 mg PO q24h (C)
Essential cofactor for transport of fatty acids into mitochondria for oxidation.
May improve fatty acid oxidation in obese cats undergoing weight loss but will not
prevent hepatic lipidosis. Used in ancillary treatment of hepatic lipidosis.
Zinc
Zinc acetate: Galzin (Gate Pharmaceuticals); many others available
To decrease Cu absorption: 100-mg elemental zinc PO q12h for 2–3 months, then 50 mg
PO q12h (D). For zinc supplementation in chronic liver disease: 1–2 mg/kg PO q12h
(D)
Induces intestinal metallothionein, which preferentially binds Cu and decreases absorption.
Zinc has antioxidant and antifibrotic effects; supports cell membrane function and
immune response.
Many zinc products are available (acetate, sulfate, gluconate). Avoid zinc methionine
if impending HE. Calculate dose on elemental zinc content. Monitor blood zinc levels:
Ideal = 200–400 mg/dl; avoid >800 mg/dl (hemolysis). Do not give concurrently with
Cu chelator (will be chelated). Nausea, vomiting, decreased appetite (less with zinc
acetate).
Vitamin C (ascorbic acid)
Many available
100–500 mg PO q24h (D&C)
Free radical scavenger; functions in converting vitamin E back to active form. Acts
as pro-oxidant in the presence of high iron, Cu levels.
Avoid in Cu-associated hepatopathy.
C, cats; Cu, copper; D, dogs; HE, hepatic encephalopathy.
*
No zinc in the 9 mg tab for cats.
•
Hepatoprotectants include a varied group of compounds (prescription drugs, nutraceuticals,
vitamins) that may protect the liver from injury caused by free radicals, bile salts,
drugs, environmental toxins, or other insults.
•
Nutraceuticals are products that have characteristics of both a nutrient and a pharmaceutical.
They are readily available through health food stores or Internet
mail-order sites, yet for many of these products there is a lack of safety and efficacy
data.
•
Lack of a regulatory mechanism for nutraceuticals is not justification for ignoring
the potential therapeutic benefits of some of these products. However, most of these
drugs have not been studied adequately in spontaneous hepatobiliary diseases of dogs
and cats to prove efficacy.
ACUTE HEPATIC FAILURE
Acute hepatic failure occurs when a sudden severe insult to the liver compromises
at least 70% to 80% of functional hepatic tissue. The clinical manifestations and
laboratory findings associated with acute hepatic failure reflect general liver failure
and are not specific for the underlying cause of injury.
Etiology
Causes of acute hepatic injury in dogs and cats include hepatotoxins, infectious or
parasitic agents, systemic or metabolic disorders, or miscellaneous causes of liver
injury (Table 71-3
). In many cases, a specific cause cannot be identified.
Table 71-3
CAUSES OF ACUTE HEPATIC INJURY IN DOGS AND CATS
Hepatotoxins
Infectious or Parasitic Agents
Drugs and Anesthetics
Viral
Acetaminophen
(D&C)
Infectious canine hepatitis (adenovirus I)
Amiodarone
(D)
Canine herpesvirus
Aspirin
(D&C)
Feline infectious peritonitis (coronavirus)
Anticonvulsants
Feline calicivirus (virulent strains)
Phenobarbital*
(D)
Phenytoin*
(D)
Bacterial
Primidone*
(D)
Cholangiohepatitis
Valproic acid*
(D)
Leptospirosis
Diazepam
(C)
Liver abscess
Aprindine
(D)
Bacillus piliformis (Tyzzer's Disease)
Azathiaprine
(D)
Salmonella spp.
Carprofen
(D)
Francisella tularensis (Tularemia)
Danazol
(D)
Glipizide
(C)
Fungal
Griseofulvin
(C)
Histoplasma
Halothane
(D)
Coccidioides
Itraconazole
(D&C)
Blastomyces
Ketoconazole
(D&C)
Others
Lomustine (CCNU)*
(D)
Macrodantin
(D)
Protozoal and Parasitic
Mebendazole
(D)
Toxoplasma
Megestrol acetate
(C)
Babesia
Methimazole
(C)
Cytauxzoon felis
Methotrexate
(D)
Dirofilaria (Postcaval syndrome)
Methoxyflurane
(D)
Mibolerone
(D)
Rickettsial
Mithramycin
(D)
Ehrlichia canis
Mitotane
(D)
Rickettsia rickettsii
Oxibendazole
(D)
Phenazopyridine
(C)
Systemic or Metabolic Disorders
Potentiated sulfonamides
(D)
Acute pancreatitis
Stanozolol
(C)
Acute hemolytic anemia
Tetracycline
(D&C)
Extraheptic infection, septicemia and endotoxemia
Thiacetarsamide
(D)
Tolbutamide
(D)
Idopathic feline hepatic lipidosis
Inflammatory bowel disease
Biologic Substances
Aflatoxin (contaminated dog food)
Traumatic, Thermal, or Hypoxic Injury
Amanita mushrooms
Blue-green algae
Abdominal trauma
Sago palms
Diaphragmatic hernia with liver entrapment
Hymenoptera toxins (hornet sting)
Heat stroke
Indigofera sp. (toxic plant)
Liver lobe torsion
Pennyroyal oil
Shock
Surgical hypotension and hypoxia
Chemicals
Carbon tetrachloride
Dimethylnitrosamine
Galactosamine
Metals (copper, iron, zinc, phosphorus)
Organochloride pesticides
Salt poisoning
Many others
*
Usually causes chronic rather than acute hepatic disease. D, dog; C, cat.
Toxin-Induced Injury
•
Hepatic injury may occur after exposure to a wide variety of industrial chemicals,
organic solvents, pesticides, heavy metals, and biologic toxins (see Table 71-3).
Exposure can be unobserved in a free-roaming animal that drinks from a contaminated
water source.
•
When hepatic necrosis is severe and widespread, rapid deterioration and death in 3
to 4 days often occur. With less extensive damage, complete recovery is possible.
Drug-Induced Injury
Drug-induced injury is a recognized cause of acute hepatic failure in dogs and cats.
The incidence of drug-induced hepatic disease is unknown but is probably underestimated.
Hepatic drug reactions are categorized as dose dependent or idiosyncratic.
Dose-Dependent Hepatotoxins
These hepatotoxins predictably damage the liver in an exposed population. The effect
is dose-related and reproducible experimentally. All members of a species are affected
at high doses. Toxicity is due to the parent drug or a consistently generated toxic
metabolite.
•
If a hepatotoxic reaction occurs, lowering the dose, rather than stopping the drug,
can be tried.
•
Examples of dose-related hepatotoxins include acetaminophen, tetracycline, stanozolol
(cats only), and possibly phenobarbital (dogs only).
Idiosyncratic Hepatotoxins
These hepatotoxins cause hepatic injury at therapeutic doses in only a few individuals
in the exposed population. These reactions are unpredictable and infrequent; most
individuals treated with the drug do not have a reaction, even at high doses. Affected
individuals appear to be unusually susceptible, possibly because they generate a unique
toxic intermediate metabolite. An immunologic response may or may not be involved.
Within susceptible individuals, toxicity may be more pronounced at higher doses. Because
of the unpredictable nature of the reaction, they can be difficult to recognize clinically.
•
If an idiosyncratic reaction occurs, the drug must be discontinued or it could result
in the death of the patient.
•
Examples include halothane and methoxyflurane, carprofen and other NSAIDs, lomustine,
mebendazole, oxibendazole, potentiated sulfonamides, and methimazole.
•
Drugs that have been incriminated as potential hepatotoxins include analgesics, anticonvulsants,
and antimicrobials (see Table 71-3). Specific clinical details regarding known drug
reactions in dogs and cats are summarized in Table 71-4
.
Table 71-4
SELECTED HEPATOTOXIC DRUG REACTIONS IN DOGS AND CATS
Drug
Species
Onset of Signs and Key Features
Hepatic Lesions
Suggested Mechanism
Comments
Analgesics
Acetaminophen (Tylenol, McNeil)
Canine and feline
Initial toxicity is cyanosis and methemoglobinemia. Acute hepatic failurea occurs
in dogs but is less likely in cats.
Centrilobular necrosis and congestion, vacuolar hepatopathy and bile stasis
Dose-related
Dose-related injury (doses exceeding 200 mg/kg in dogs and 120 mg/kg in cats). Treatment:
N-acetylcysteine (140 mg/kg IV or PO initially, then 70 mg/kg IV or PO q6h for 36
hours) and ascorbic acid (30 mg/kg q6h for 36 hours). SAMe (20 mg/kg PO q24h) can
also serve as a glutathione source. Cimetidine (5–10 mg/kg IV q8–12h for at least
3 days) inhibits hepatic P-450 enzymes and prevents further conversion of acetaminophen
to toxic product.
Carprofen (Rimadyl, Pfizer)
Canine
Acute hepatic failurea within 5–30 days after starting drug. Labrador retrievers may
be at increased risk.
Hepatocellular necrosis, ballooning degeneration, cholestasis
Idiosyncratic
Evaluate liver and kidney function prior to starting treatment. Concurrent renal tubular
necrosis and glucosuria may occur. Rapid recovery in Labradors after stopping drug
and giving supportive care. A 50% mortality rate in non-Labrador breeds. Whether dogs
with carprofen hepatotoxicity can be safely switched to another NSAID without experiencing
a hepatic reaction is unknown.
Anticonvulsants
Phenobarbital
Canine
Chronic liver disease and cirrhosis. Anorexia, lethargy, weight loss, sedation, PU/PD,
icterus, ascites, encephalopathy. ↑ ALP, ↑ ALT, ↑ GGT, ↑ total bilirubin, ↑ SBA, hypoalbuminemia,
hypocholesterolemia, coagulopathy.
Hepatocellular hypertrophy and vacuolar hepatopathy (early); bridging portal fibrosis,
nodular regeneration, biliary hyperplasia, mild inflammatory infiltrates
Dose-related?
Phenobarbital causes chronic hepatic disease and cirrhosis when given at high doses
(serum concentrations >40 μg/ml) or for long periods (months to years). Dogs with
phenobarbital hepatotoxicity may improve when dosage is decreased to therapeutic range
as determined by serum phenobarbital levels or when potassium bromide therapy is substituted
for phenobarbital. Incidence of chronic hepatic disease from long-term anticonvulsant
therapy is approximately 6–14%. Hepatocutaneous syndrome has also been described in
dogs on long-term phenobarbital therapy.
Diazepam
Feline
Acute hepatic failurea within 4–13 days of starting oral drug administration.
Diffuse hepatic necrosis
Idiosyncratic
Most cats with hepatotoxicity die within 15 days of initial administration of drug.
In cats treated with oral diazepam, monitor baseline liver enzymes before and within
5 days after starting therapy; if enzymes are increased, discontinue drug and give
supportive therapy.
Antimicrobials
Ketoconazole (Nizoral, Janssen Pharmaceuticals)
Canine and feline
Asymptomatic with ↑ALT, ↑ ALP, or rarely acute hepatic failure.a
Not characterized
Idiosyncraticb
May be dose-related phenomenon: >40 mg/kg/day in dogs. In humans, asymptomatic serum
enzyme elevations are considered harmless and enzymes usually return to normal despite
continued therapy. Therapy should be discontinued if ↑ ALT >3 times normal or if clinical
signs or jaundice occur. Recovery is usually uneventful.
Itraconazole (Sporanox, Janssen Pharmaceuticals)
Canine and feline
Asymptomatic with ↑ ALT or anorexia.
Not characterized
Dose-related?
Hepatotoxicity most likely at higher doses. Therapy should be stopped if ↑ ALT >3
times normal or if anorexia occurs. When appetite returns, can reinstitute itraconazole
at half the original dose.
Drug
Species
Onset of Signs and Key Features
Hepatic Lesions
Suggested Mechanism
Comments
Tetracycline
Canine and feline
Not characterized.
Vacuolar hepatopathy
Dose-related
Experimental hepatic injury induced by high doses given IV. Avoid in animals with
PSS. Unlikely to be clinically important as a hepatotoxin. Not clear whether doxycycline
has same effect on liver.
Potentiated sulfonamidesc
Canine
Acute hepatic failurea 5–36 days after starting therapy.
Diffuse necrosis or periportal hepatitis and intrahepatic cholestasis
Idiosyncratic
Multiple exposures may predispose to reaction, but previous exposure to sulfonamides
is not required. Dogs with intrahepatic cholestasis recover rapidly after drug withdrawal.
High mortality with diffuse hepatic necrosis. Consider treatment with N-acetylcysteine
and vitamin C or SAMe to promote detoxification of toxic intermediate, nitroso-SMX.
Steroids
Glucocorticoids (various types)
Canine
Chronic hepatopathy, but hepatic failure rare. Signs indicative of hypercortisolism
(e.g., PU/PD, polyphagia, hepatomegaly, and lethargy). ↑ ALP, usually with induction
of steroid isoenzyme of ALP. Mild ↑ ALT; normal total bilirubin. SBA within normal
limits or mildly ↑ (<60 μmol/L).
Centrilobular vacuolization due to glycogen accumulation
Dose-related but considerable individual variation
Lesions reversible after treatment is discontinued. Does not occur in cats.
Stanozolol (Winstrol-V, Pharmacia and Upjohn)
Feline
Increased ALT and hyperbilirubinemia; anorexia.
Hepatic lipidosis
Dose-related
Most cats recover after discontinuing stanozolol and after supportive care.
Miscellaneous
Methimazole (Tapazole, Lilly)
Feline
Acute hepatic failurea within 2 months of starting therapy.
Necrosis and cholestasis
Idiosyncratic
Clinical signs resolve within 7 days of stopping therapy. Biochemical resolution by
45 days.
Lomustine (CCNU) (CeeNu, Bristol-Meyers Squibb)
Canine
Chronic liver disease with decreased appetite, weight loss, PU/PD, vomiting, ascites
and ↑ ALT, ↑ ALP, hypoalbuminemia.
Hepatocellular vacuolization, mild to moderate periportal inflammation, fibrosis,
hemosiderin-laden Kupffer cells
Idiosyncratic
Median time to detection of hepatic disease from last dose of CCNU was 11 weeks (range
of 2–49 weeks). Cumulative drug doses and number of doses tends to be higher in dogs
that develop hepatotoxicity than in those that do not. Majority of affected dogs die
of progressive chronic liver disease.
ALP, alkaline phosphatase; ALT, alanine aminotransferase; GGT, gamma-glutamyltransferase;
NSAID, nonsteroidal anti-inflammatory drug; PSS, portosystemic shunt; PU/PD, polyuria/polydipsia;
SAMe, S-adenosylmethionine; SBA, serum bile acid.
a
Typical manifestations of acute hepatic failure include anorexia, depression, vomiting,
and jaundice accompanied by ↑ ALT, ↑ ALP, and ↑ serum bilirubin.
b
For drug reaction accompanied by clinical signs and hyperbilirubinemia.
c
Includes trimethoprim-sulfadiazine, trimethoprim-sulfamethoxazole, and ormetoprim-sulfadimethoxine.
Key Point
Dose-dependent hepatotoxic drugs cause predictable liver injury in all animals of
a species, especially at high or excessive doses, whereas idiosyncratic hepatotoxic
drugs affect only certain uniquely susceptible animals, even at low or therapeutic
doses.
Infectious Agents
Infectious causes of acute hepatic injury (see Table 71-3) include leptospirosis (see
Chapter 19), toxoplasmosis (see Chapter 21), histoplasmosis (see Chapter 20), FIP
virus (see Chapter 10), and infectious canine hepatitis virus (see Chapter 16).
Miscellaneous Systemic Diseases
Hepatic injury can occur secondary to various systemic conditions, including those
discussed below.
Hemolytic Anemia
Hemolysis, especially immune-mediated hemolytic anemia in dogs, can be complicated
by centrilobular necrosis attributed to acute hepatocellular hypoxia or DIC-induced
sinusoidal thrombosis. The hepatic injury generally resolves with resolution of the
anemic crisis.
Anesthesia, Surgical Hypotension, Hypoxia, and Shock
Conditions that decrease liver blood flow can lead to hypoxia and hepatic damage.
In the postoperative
period it is difficult to differentiate hepatic damage or jaundice caused by the hypoxic
effects of anesthesia and surgery from other potential causes, such as toxic injury
induced by anesthetic agents (e.g., halothane or methoxyflurane) and other drugs,
postoperative infections, endotoxemia, or a combination of these factors.
Acute Pancreatitis
Pancreatitis in dogs and cats often is characterized by increased serum concentrations
of liver enzymes resulting from secondary hepatic injury from released enzymes and
inflammatory cytokines (see Chapter 73). Hepatic lesions usually resolve with resolution
of the pancreatitis and require no special treatment. Less commonly, jaundice results
from partial to complete obstruction of the common bile duct associated with peripancreatic
inflammation, pancreatic abscess, or pancreatic healing by fibrosis. Surgical intervention
to relieve biliary obstruction is indicated in animals with pancreatic abscess or
if jaundice persists after resolution of acute pancreatitis, which usually indicates
pancreatic fibrosis as a cause of biliary obstruction (see Chapter 73).
Extrahepatic Bacterial Infections
Septicemia and endotoxemia associated with infections such as pneumonia, pyometra,
peritonitis, and abscesses can cause jaundice, mild to moderate increases in liver
enzyme activity (especially ALP), and increased SBA concentration. Liver biopsy reveals
intrahepatic cholestasis without significant necrosis or inflammation. The hepatic
injury resolves when the infection is controlled.
Clinical Signs
Clinical signs of acute hepatic failure often are nonspecific and overlap signs of
disorders of other body systems. Clinical signs reflect general hepatic dysfunction
rather than the specific underlying cause. Signs of extrahepatic or multisystemic
disease often provide important diagnostic clues when hepatic injury occurs secondary
to acute pancreatitis, hemolytic disease, septicemia or endotoxemia, and many infectious
diseases.
•
Acute onset of anorexia, lethargy, vomiting, and diarrhea are the most common presenting
signs of acute hepatic failure.
•
Other potential findings include PU/PD, jaundice, excessive bleeding, and HE.
Diagnosis
•
When acute hepatic failure is diagnosed, attempt to identify the underlying cause
with a complete history, ancillary diagnostic testing, and if indicated, a liver biopsy.
In many cases, a specific cause cannot be identified.
•
When acute hepatic failure is accompanied by jaundice, consider diseases of the extrahepatic
biliary tract, such as biliary obstruction and rupture. Surgical intervention may
provide both diagnostic and therapeutic benefit.
History
Attempt to document recent or potential exposure to any drug, toxin, or infectious
disease, especially those listed in Table 71-3.
•
Suspect a drug- or toxin-induced cause of acute hepatic failure when clinical and
biochemical evidence of acute hepatic dysfunction is associated with recent exposure
to a potential hepatotoxin.
•
Consider toxin-induced injury even in the absence of known exposure to toxins, because
potential hepatotoxins can be present in contaminated dog food (aflatoxins), pond
water (cyanobacteria, or “blue-green algae”), and many other unobserved sources.
•
Although numerous drugs have been incriminated (see Table 71-3, Table 71-4) remember
that an idiosyncratic reaction can occur with any drug. With most drug- and toxin-induced
disorders, the diagnosis is presumptive and cannot be proved.
•
To confirm the diagnosis, discontinue the drug and observe for clinical improvement,
which usually occurs within several weeks, even after chronic drug administration.
Recurrence of hepatic damage after a challenge dose of the same drug (or inadvertent
re-exposure) supports the diagnosis of drug-induced hepatotoxicity. Note: This is
not recommended as a diagnostic procedure because it is potentially dangerous, especially
with a drug that causes hepatic necrosis.
•
Determine if there is a history of recent surgical or anesthetic procedures that may
be associated with drug- or hypoxia-related hepatic damage.
•
Evaluate the animal's vaccination status for infectious diseases that can involve
the liver, such as leptospirosis and infectious canine hepatitis.
•
Determine if there are any subtle chronic signs of illness that suggest that the underlying
liver disease may be chronic rather than acute and that the current illness may be
exacerbation or decompensation of chronic liver disease.
Physical Examination
Physical findings often reflect general hepatic dysfunction rather than the specific
etiology (see previous discussion of physical examination findings under “Diagnostic
Strategy for Liver Disease”).
•
Hepatodynia may occur with any cause of acute hepatic injury that results in swelling
and stretching of the liver capsule.
•
Findings of weight loss and ascites are indicative of a chronic rather than an acute
process.
•
Signs of extrahepatic or multisystemic disease may be important clues when liver injury
occurs secondary to systemic disorders.
•
For example, fever may be present with infectious causes of hepatic injury such as
leptospirosis, infectious canine hepatitis, bacterial cholangitis, liver abscess,
systemic mycoses, and extrahepatic infections that secondarily involve the liver.
•
Fever and acute abdominal pain are presenting signs of acute pancreatitis but can
also occur with cholangitis and hepatic abscess.
•
When jaundice is accompanied by pallor, consider immune hemolytic anemia.
Laboratory Evaluation
Acute hepatotoxicity frequently is associated with abnormal serum biochemical analyses,
liver function tests, and urinalysis.
•
Because diffuse hepatic necrosis is the most common lesion associated with acute hepatic
failure, increased ALT activity is the most consistent finding, and values are often
markedly increased. Increased ALP activity may also occur.
•
Other potential findings include hyperbilirubinemia, increased SBA concentrations,
hypoglycemia, hyperammonemia, and coagulopathy. Hypoalbuminemia usually suggests chronic
rather than acute liver disease.
•
Some hepatotoxins (e.g., carprofen) and infectious agents (e.g., leptospirosis) may
concurrently damage the kidneys; thus, biochemical evidence of concomitant renal failure
may be present.
•
An inflammatory CBC suggests possible acute pancreatitis or underlying infectious
disease. Evaluate serum amylase and lipase (dogs) and pancreatic lipase immunoreactivity
(PLI) (dogs and cats) to diagnose acute pancreatitis.
Abdominal Imaging
•
Liver size on abdominal radiographs usually is normal to increased unless massive
hepatic necrosis causes parenchymal collapse and microhepatica.
Key Point
A small liver generally suggests chronic rather than acute hepatic disease.
•
Additional radiographic and ultrasonographic findings may be noted, depending on the
underlying disorder (Table 71-5
).
Table 71-5
ANCILLARY DIAGNOSTIC EVALUATIONS FOR HEPATOBILIARY DISEASE
Diagnostic Evaluation
Intended Diagnosis (Rule Out)
Bacterial cultures
Liver, gallbladder, bile
Bacterial cholangiohepatitis, cholecystitis, hepatic abscess
Blood, urine, infected tissues
Extrahepatic infections and sepsis
Serologic tests (antibody titers)
Leptospirosis
Mycoses (histoplasmosis, coccidioidomycosis, blastomycosis)
Toxoplasmosis
Feline infectious peritonitis
Bartonellosis
Other infectious diseases
Microfilaria exam
Heartworm disease
Heartworm antigen or antibody tests
Heartworm disease
Fecal sedimentation (formalin-ether technique)
Liver fluke infection
Serum amylase and lipase
Acute pancreatitis in dogs
Serum pancreatic lipase immunoreactivity
Pancreatitis in cats and dogs
Serum T4
Feline hyperthyroidism
Coombs' test
Immune hemolytic anemia
Lymph node aspiration cytology
Mycoses
Lymphoma
Hepatic fine-needle aspiration cytology
Infectious agents (e.g., mycoses)
Neoplasia (e.g., lymphoma)
Feline hepatic lipidosis
Hepatic abscess (ultrasound guided)
Abdominocentesis
Ruptured biliary tract
Feline infectious peritonitis
Neoplasia
Thoracic radiography
Mycoses
Toxoplasmosis
Heartworm disease
Metastatic neoplasia
Diaphragmatic hernia
Abdominal radiography
Hepatic abscesses
Emphysematous cholecystitis
Cholelithiasis
Pancreatitis
Abdominal ultrasonography
Focal and diffuse hepatic parenchymal abnormalities
Biliary or gallbladder disease
Portosystemic shunt(s)
Hepatic arteriovenous fistulas
Pancreatic disease
Diaphragmatic hernia
Angiography
Portogram
Portosystemic shunt(s)
Portal vein obstruction
Hepatic venography
Obstruction of caudal vena cava and hepatic veins
Celiac arteriography
Hepatic arteriovenous fistulas
Nuclear imaging
Portal scintigraphy
Portosystemic shunting
Modified from Johnson SE: Diseases of the liver. In Ettinger SJ, Feldman EC (eds):
Textbook of Veterinary Internal Medicine, vol. 2, 4th ed. Philadelphia: WB Saunders,
1995, p 1316.
Liver Biopsy
Perform a liver biopsy when the cause of acute hepatic failure is not suggested by
preliminary laboratory evaluation.
•
Histopathologic examination of hepatic tissue can help establish the cause and distinguish
between acute and chronic liver disease. Diffuse hepatic necrosis is the histologic
lesion most consistently associated with acute hepatic failure.
•
If overt bleeding is present, liver biopsy may be contraindicated.
Ancillary Diagnostic Procedures
Perform ancillary diagnostic procedures (see Table 71-5) to diagnose underlying causes
of acute hepatic failure.
Treatment
•
Management of the patient with acute hepatic failure is first directed toward supportive
therapy (see Table 71-1). In many cases, even though the cause remains unidentified
or specific therapy is unavailable, supportive care alone may allow adequate time
for hepatic regeneration to occur.
•
Maintenance of fluid, electrolyte, and acid-base balance is the cornerstone of supportive
therapy (see Chapter 5).
•
Prevent or control complications such as hypoglycemia, HE, coagulopathy, and endotoxemia
(see “Principles of Treatment for Liver Disease”).
•
Whenever possible, institute specific treatment for the underlying cause; for example,
administer injectable penicillin or amoxicillin for treatment of possible leptospirosis.
Discontinue use of a suspect drug to prevent further hepatic injury and observe for
clinical improvement.
•
With the exception of acetylcysteine for acetaminophen toxicity and milk thistle (silybin)
for Amanita mushroom poisoning, no specific antidotes are available for drug- or toxin-induced
hepatic injury. However, hepatoprotectants (Table 71-2) may be helpful in treating
hepatotoxicity but have not been adequately evaluated.
INFECTIOUS AND PARASITIC HEPATIC DISEASE
The liver can be involved in many systemic infections (Table 71-6
). In some disorders, such as leptospirosis and infectious canine hepatitis, the liver
is a target organ, and evidence of liver failure dominates the clinical presentation.
In other infections, such as many of the systemic protozoal infections, the liver
is involved as a result of widespread invasion of organs with a large mononuclear
phagocyte population, such as the spleen, lymph nodes, and bone marrow (see Table
71-6). Signs of hepatic dysfunction may or may not be present and may be overshadowed
by more obvious extrahepatic involvement. Liver cytology and biopsy can be diagnostically
useful for identification of these organisms.
Table 71-6
INFECTIOUS DISEASES WITH POTENTIAL HEPATOBILIARY INVOLVEMENT*
Disease
Agent
Refer To
Viral
Infectious canine hepatitis
Canine adenovirus I
Chapter 16
Systemic neonatal herpesvirus
Canine herpesvirus
Chapter 16
Canine acidophil cell hepatitis
Unknown
Chapter 16
Feline infectious peritonitis
Feline coronavirus
Chapter 10
Feline systemic hemorrhagic-like febrile disease
Feline calicivirus (virulent strains)
Chapter 11
Bacterial
Leptospirosis
Leptospira interrogans sensu strictu
Chapter 19
Acute hepatic failure
Serovars icterohaemorrhagiae, canicola, autumnalis, pomona, bratislava, bataviae,
hardjo, and Leptospira kirshneri serovar grippotyphosa
Chronic hepatitis
Serovars grippotyphosa and australis
Tyzzer's disease
Bacillus piliformis
Chapter 69
Nocardiosis
Nocardia species
Chapter 19
Actinomycosis
Actinomyces species
Chapter 19
Tuberculosis
Mycobacterium tuberculosis, M. bovis, M. avium
Chapter 19
Salmonellosis
Salmonella typhimurium
Chapter 69
Brucellosis
Brucella canis
Chapter 19
Bartonellosis (dogs)
Bartonella henselae, B. clarridgeiae
Chapter 19
Hepatic abscess
Gram-negative bacteria (especially Escherichia coli), anaerobes, mixed infections
common, Staphylococcus species (puppies)
Cholangitis/cholangiohepatitis
Gram-negative bacteria (especially Escherichia coli), anaerobes
Cholecystitis
Gram-negative bacteria (especially Escherichia coli), Campylobacter jejuni, Clostridium
species
Yersiniosis
Yersinia pestis
Chapter 19
Tularemia
Francisella tularensis
Chapter 19
Fungal
Histoplasmosis
Histoplasma capsulatum
Chapter 20
Blastomycosis
Blastomyces dermatitidis
Chapter 20
Coccidioidomycosis
Coccidioides immitis
Chapter 20
Aspergillosis
Aspergillus terreus
Chapter 20
Others
Protozoal
Toxoplasmosis
Toxoplasma gondii
Chapter 21
Babesiosis
Babesia canis, B. gibsoni
Chapters 21 & 22
Cytauxzoonosis
Cytauxzoon felis
Chapters 21 & 22
Hepatozoonosis
Hepatozoon canis
Chapter 21
Leishmaniasis
Leishmania species
Chapter 21
Encephalitozoonosis
Encephalitozoon cuniculi
Chapter 21
Rickettsial
Ehrlichiosis
Ehrlichia species
Chapter 17
Rocky Mountain spotted fever
Rickettsia rickettsii
Chapter 17
Algal
Protothecosis
Prototheca species
Chapter 69
Parasitic
Canine schistosomiasis
Heterobilharzia americana
Visceral larval migrans
Toxocara canis
Liver flukes
Platynosomum concinnum (feline)
Amphimerus pseudofelineus (feline)
Canine hepatic capillariasis
Capillaria hepatica
Canine hepatic alveolar echinococcosis
Echinococcus multilocularis
*
Not necessarily associated with clinical hepatobiliary disease.
Systemic infections are covered in detail elsewhere in this book. Infections localized
to the hepatobiliary tract are covered in greater detail here.
Infectious Causes of Liver Disease
Several specific infectious diseases that involve the liver are listed in Table 71-6
and are discussed elsewhere in this book, including leptospirosis (see Chapter 19),
ehrlichiosis and rickettsial diseases (Chapter 17), toxoplasmosis (see Chapter 21),
systemic mycoses (see Chapter 20), FIP virus (see Chapter 10), and infectious canine
hepatitis virus (see Chapter 16).
Hepatic Abscess
Hepatic abscesses from bacterial infection of the liver occur uncommonly in dogs and
cats. Abscesses may form as a solitary large mass, multiple small masses scattered
throughout the liver, or microabscesses that are only detected histologically.
Etiology
•
Potential sources of bacteria include hematogenous spread, translocation of intestinal
bacteria into the portal blood, ascension via bile ducts, penetrating abdominal and
caudal thoracic wounds, and direct extension from local suppurative diseases. Umbilical
infections are the most common cause of hepatic abscesses in puppies (Staphylococcus)
and kittens (Streptococcus).
•
Escherichia coli and anaerobic bacteria are most commonly identified; mixed bacterial
infections are common.
•
Hypoxia of hepatic tissue caused by hepatic neoplasia, liver lobe torsion, or trauma
may predispose the patient to abscess formation, because small numbers of anaerobes
(e.g., Clostridium spp.) normally are present in the liver and can proliferate under
these conditions.
•
Systemic diseases that are associated with immunosuppression (e.g., feline leukemia
virus and feline immunodeficiency virus) or that predispose the patient to infection
(e.g., diabetes mellitus) may predispose the patient to hepatic abscesses.
•
Systemic infections (urinary tract infection, pneumonia), pancreatitis, gallbladder
rupture, and previous surgical liver biopsy have also been associated with hepatic
abscesses.
Clinical Signs
•
Signs are attributed to sepsis, inflammation, and hepatic dysfunction and include
anorexia, lethargy, fever, vomiting, and diarrhea.
•
Rupture of a hepatic abscess leads rapidly to peritonitis, septic shock, and death.
Diagnosis
•
Physical examination findings are often vague but may include depression, fever, hepatomegaly,
abdominal tenderness, and abdominal effusion.
•
Potential laboratory findings include neutrophilia with a left shift (or neutropenia
and degenerative left shift if rupture occurs), thrombocytopenia, markedly increased
ALT and ALP activity (although they may be in the normal range), hyperglobulinemia,
hyperbilirubinemia, hypoglycemia, and septic suppurative abdominal effusion. Increased
ALT and ALP activity occur in less than 50% of cats with hepatic abscesses.
•
Radiolucent areas may be seen on abdominal radiographs when gas-producing organisms
are involved. Ultrasonography may reveal one or more parenchymal abscess cavities.
They appear as poorly echogenic lesions that may be round, oval, or irregular in shape.
Ultrasound-guided FNA for cytology and culture may be diagnostic.
•
Diagnosis often is established at exploratory laparotomy to determine the cause of
septic peritonitis.
•
Perform aerobic and anaerobic cultures of abdominal effusion, blood, and hepatic tissue.
Treatment
•
Treatment of large, unifocal hepatic abscesses requires surgical excision of the affected
liver lobe (see Chapter 72).
•
Ultrasound-guided percutaneous abscess drainage has been advocated for medical management
of single lesions. No complications were observed in one series of cases.
•
Initiate broad-spectrum antibiotics such as intravenous penicillin combined with an
aminoglycoside (e.g., gentamicin or amikacin) while awaiting culture results. Monitor
renal function during aminoglycoside therapy, and base further therapy on results
of sensitivity testing. Give long-term antibiotic therapy (but not aminoglycosides)
for at least 6 to 8 weeks.
•
The mortality rate was high (79%) in a recent series of cats with hepatic abscesses
as compared with dogs (50%).
Liver Fluke Infection
Liver fluke infection is uncommon in cats and rare in dogs. Infection usually is asymptomatic
but may cause clinical biliary tract disease when associated with marked biliary fibrosis,
cholangitis, cholangiohepatitis, or extrahepatic bile duct obstruction. Cholangiocarcinoma
has been reported in cats chronically infected with Platynosomum concinnum.
Etiology
•
P. concinnum (Platynosomum fastosum) is the most important liver fluke in cats and
is found in tropical and subtropical geographic areas, including Hawaii, Florida,
and the Caribbean. In endemic areas, the prevalence of infection is high.
•
Other liver flukes that have been identified in cats include Amphimerus pseudofelineus
(Opisthorchis pseudofelineus), Opisthorchis tenuicollis, Opisthorchis sinensis, and
Metorchis conjunctus (Metorchis complexus).
•
Liver flukes require two intermediate hosts for their life cycle. Adult flukes reside
in the gallbladder and bile ducts. Embryonated eggs are shed in the feces and ingested
by a snail, the first intermediate host for all liver flukes. The house gecko, skink,
lizard, and Bufo toad are second intermediate hosts for P. concinnum; fish are second
intermediate hosts for the other species of flukes.
Clinical Signs
•
Most infected cats are asymptomatic for liver fluke infection.
•
Liver fluke infection occasionally is associated with anorexia, weight loss, diarrhea,
vomiting, jaundice, hepatomegaly, abdominal distention, and death.
Diagnosis
•
Operculated fluke eggs can be identified in feces by a formalin-ether technique (a
sedimentation procedure). Routine methods for flotation do not consistently identify
eggs. With complete bile duct obstruction, no eggs will be passed in the feces. Eggs
may be identified on cytologic examination of the bile.
•
Other laboratory findings are inconsistent and often unremarkable. Eosinophilia, hyperbilirubinemia,
and increased serum ALP and ALT activity are sometimes detected.
•
A possible relationship between positive feline immunodeficiency virus status and
infection with A. pseudofelineus has been suggested.
•
At laparotomy or necropsy, the bile ducts and gallbladder may be distended and thick
walled and may contain inspissated bile and small (<12 mm long) adult flukes. The
liver frequently is enlarged. In many cases, no visible abnormalities are present.
Treatment
Minimal information is available about treatment of liver flukes.
•
Praziquantel (Droncit), 40 mg/kg, given orally or parenterally once a day for 3 consecutive
days, or fenbendazole (Panacur) 50 mg/kg PO q24 for 10 to 14 days, has been suggested.
•
Drugs used unsuccessfully include mebendazole, levamisole, thiabendazole, diamphenethide,
and rafoxanide. At least one follow-up fecal examination by formalin-ether sedimentation
should be performed 30 days following treatment.
•
Manage complications such as biliary obstruction and secondary bacterial cholangitis
or cholangiohepatitis as described elsewhere in this chapter.
FELINE HEPATIC LIPIDOSIS
Hepatic lipidosis is an excessive accumulation of triglyceride in the liver that occurs
when there is an imbalance between the rate of deposition and the rate of mobilization
of fat from the liver. It is the most common liver disease in cats and is associated
with severe intrahepatic cholestasis and hepatic failure. Mortality is high if the
disorder is untreated.
Etiology
General mechanisms of hepatic lipidosis include nutritional, metabolic, hormonal,
toxic, and hypoxic liver injury. Diabetes mellitus is a well-recognized and easily
diagnosed cause of hepatic lipidosis. Drug- (tetracycline, stanozolol) or toxin-induced
injury can also cause histologic lesions of lipidosis. Hepatic lipidosis often occurs
secondary to other systemic disorders associated with anorexia and a catabolic state,
especially cholangitis, pancreatitis, inflammatory bowel disease (IBD), and systemic
neoplasia. The term idiopathic hepatic lipidosis is used when no other underlying
causative disease is identified.
The following mechanisms may be important in the development of idiopathic hepatic
lipidosis:
•
Cats have higher nutritional requirements for protein, essential amino acids, and
essential fatty acids than dogs.
•
Systemically ill cats have a propensity for accumulating fat in their hepatocytes.
•
Profound anorexia and stress may be associated with hormonal (catecholamines, other)
alterations that influence fat metabolism and predispose the patient to peripheral
fat mobilization and hepatic fat uptake.
•
Obese cats do not seem to be able to adapt to metabolism of fat for energy during
periods of starvation.
Key Point
Persistent anorexia and rapid weight loss are hallmarks of severe hepatic lipidosis.
It most commonly develops in overweight cats that experience prolonged (usually >2
weeks) inappetence, sometimes triggered by a stressful event.
•
The exact mechanism or biochemical aberration in cats with hepatic lipidosis is unknown.
However, there appears to be an imbalance in the mobilization of peripheral fat, the
hepatic use of fatty acids for energy, and the hepatic dispersal of triglycerides.
Clinical Signs
•
Hepatic lipidosis is a disease of middle-aged or older cats without breed or gender
predilection. Many affected cats are obese prior to the onset of disease.
•
Prolonged anorexia, often several weeks in duration, is the most consistent clinical
sign.
•
Other findings include lethargy, vomiting, constipation or diarrhea, and weight loss.
Weight loss can be dramatic and may exceed 25% of the previous weight.
•
Overt signs of HE (hypersalivation, severe depression, stupor) are uncommon.
•
Overt bleeding occurs in 20% of cases.
Diagnosis
Clinical findings and laboratory evaluation in cats with hepatic lipidosis suggest
hepatic disease, but liver biopsy is required to distinguish hepatic lipidosis from
other causes of hepatic disease such as cholangitis, FIP, and neoplasia. When hepatic
lipidosis occurs secondary to another disorder, additional testing is required to
identify the primary disease (e.g., pancreatic lipase immunoreactivity for pancreatitis
or GI endoscopy and biopsy for IBD).
History
The history may reveal precipitating causes of anorexia such as stressful events (e.g.,
boarding, surgery, or change in living arrangements), a diet change for weight reduction,
or non-hepatic diseases associated with anorexia.
Physical Examination
•
Findings include hepatomegaly, jaundice, muscle wasting, seborrhea, and pallor.
•
Ventroflexion of the head and neck occurs in some cats and may represent muscle weakness
associated with electrolyte imbalances (hypokalemia, hypophosphatemia) or thiamine
deficiency.
Laboratory Evaluation
•
Hematologic findings are nonspecific and include a nonregenerative, normocytic, normochromic
anemia with poikilocytosis and mature neutrophilia and lymphopenia (stress response).
Hemolysis may occur secondary to hypophosphatemia or Heinz bodies.
•
Serum ALP, ALT, and AST activities; FSBA and PPSBA concentrations; and total serum
bilirubin concentration usually are increased. Increases in liver enzymes precede
increases in total bilirubin and bile acids.
Key Point
Serum ALP activity is generally higher in cats with lipidosis than with other hepatic
diseases. Serum GGT activity, which usually parallels or exceeds serum ALP activity
in most feline hepatic diseases, is normal or only mildly increased in hepatic lipidosis.
•
Other potential findings include hypokalemia, hyperammonemia, hypoalbuminemia, and
decreased BUN. Many affected cats have abnormal coagulation tests, especially PIVKA
values and hypofibrinogenemia. In one study, PIVKA values improved in 50% of the cats
treated with vitamin K1, suggesting acquired vitamin K deficiency.
•
Consider serum pancreatic lipase immunoreactivity to evaluate for concurrent pancreatitis.
•
Consider serum cobalamin (B12) levels if an underlying intestinal disorder suspected.
Radiography and Ultrasonography
•
Radiographically, the liver is normal to increased in size.
•
Ultrasonographic findings include hepatomegaly and diffuse hyperechogenicity of the
liver. Ascites is rare. Evaluate for concurrent pancreatitis or other disorders causing
secondary hepatic lipidosis.
Fine-Needle Aspiration Cytology
•
FNA cytology is a less invasive alternative to liver biopsy that can provide similar
information. Results of FNA cytology occasionally can be misleading because the small
sample size may not be representative of the pathologic process in the liver.
•
Correct coagulopathy with vitamin K1 prior to performing liver aspirate or liver biopsy.
•
On cytologic evaluation, hepatocytes are foamy and vacuolated, and inflammatory cells
are absent.
Liver Biopsy
Liver biopsy is required for definitive diagnosis but is not routinely performed unless
there is failure to respond to appropriate therapy or a high level of suspicion of
another primary hepatic disorder.
•
Grossly, the liver is enlarged, yellow, greasy, and friable with rounded edges. Biopsy
specimens usually float in formalin.
•
On routine H&E staining, there is severe vacuolization of hepatocytes (>50% of acinar
unit involved).
•
Oil red O stain performed on formalin-fixed (non–paraffin-embedded) frozen tissue
can confirm excess fat in the vacuoles.
•
Inflammation or necrosis usually is absent.
Treatment
Because of lack of information regarding the underlying cause, treatment is primarily
supportive. The greatest success has been with aggressive nutritional support. During
initial stabilization, correct dehydration, electrolyte imbalances, coagulopathies
and any complications of liver failure. A nasogastric tube can be placed for short-term
nutritional support. CliniCare (Abbott Veterinary Diets) can be used initially through
the nasogastric tube while stabilizing the patient prior to anesthesia for a longer-term
feeding tube (gastrostomy or esophagostomy tube).
Initial Fluid Therapy
•
Use intravenous fluid therapy with a balanced electrolyte solution supplemented with
potassium chloride in the initial stages of treatment (see Table 71-1). Hepatic lactate
metabolism may be impaired in cats with hepatic lipidosis; thus, avoid lactated Ringer's
solution.
•
If hypokalemia persists despite supplementation, evaluate serum magnesium concentration
to see if hypomagnesemia is the cause of refractory hypokalemia.
Key Point
Avoid dextrose supplementation unless hypoglycemia is documented, because glucose
may promote hepatic lipid accumulation if caloric needs are not being adequately met.
•
Monitor serum phosphorus concentration and treat with IV potassium phosphate if levels
decline to <2 mg/dl (see Chapter 5). Hemolysis may occur secondary to severe hypophosphatemia.
•
Abnormal blood coagulation test results and excess bleeding occasionally respond to
vitamin K1 therapy, suggesting severe cholestasis and vitamin K malabsorption (see
Table 71-1). Consider fresh blood transfusion as needed for management of anemia.
•
Treat HE as described in Table 71-1, using a low-protein diet, lactulose, and amoxicillin,
neomycin, or metronidazole.
•
Consider antibiotic therapy with amoxicillin to prevent infection secondary to compromised
hepatic clearance of enteric organisms. Avoid tetracycline because it can predispose
the patient to hepatic lipid accumulation.
Nutritional Therapy
•
Provide the daily caloric requirement (40-60 kcal/kg of body weight per day) via nasogastric,
esophagostomy, or gastrostomy tube. An endoscopically placed gastrostomy tube is preferable
because long-term nutritional therapy (at least 3-6 weeks) is necessary in most cases.
Nasogastric tubes are adequate for short-term management and are preferable to force-feeding.
Techniques for placement of indwelling feeding tubes are described in Chapter 3.
•
Feed Maximum Calorie (Iams), Prescription Diet a/d (Hill's Pet Nutrition), or other
complete and balanced feline diet that can be delivered through a tube in small feedings.
•
Initially, give one-fourth to one-half of the daily dietary caloric requirement through
the tube, divided into 4 to 6 feedings per day. Gradually increase the total amount
fed over 3 to 4 days until maintenance requirements are achieved.
•
Use a restricted-protein diet only if hyperammonemia or overt signs of HE occur.
Key Point
Do not rely on appetite stimulant medications, because they rarely achieve the consistent
caloric intake required for effective reversal of lipidosis. Avoid diazepam, in particular,
because it has been associated with idiosyncratic hepatic necrosis in cats.
•
If vomiting or delayed gastric emptying is a problem, give metoclopramide (0.4 mg/kg
SC q8h, 30 minutes before feeding), or feed a liquid enteral diet by constant rate
infusion into the feeding tube. Dilution of the diet with water may also improve tolerance.
Dietary Supplements
Numerous vitamins and supplements have been empirically recommended in the treatment
of idiopathic hepatic lipidosis, but further controlled studies are needed to determine
clinical usefulness. Consider each of the following:
•
B complex vitamins
added to the fluids (1-2 ml).
•
Cobalamin (B12)
, 250μg SC initially while awaiting serum cobalamin levels. If decreased serum cobalamin
is documented (usually indicating primary intestinal disease), continue it long term
(see Chapter 69).
•
Thiamine
(if severe ventroflexion of neck), at a dosage of 50 to 100 mg PO q24h, for 1 week
(or added to IV fluids).
•
L-Carnitine
, 250 to 500 mg PO q24h, as an essential cofactor for fatty acid oxidation (for relative
carnitine deficiency).
•
Taurine
, at a dosage of 250 to 500 mg PO q24h, for the initial 7 to 10 days. Plasma taurine
is decreased in many cats with hepatic lipidosis, and taurine is required for bile
acid conjugation.
•
Vitamin E
(water-soluble form), 50 to 100 units total dose per cat PO q24h, as an antioxidant.
•
S-adenosylmethionine (SAMe)
(Denosyl SD, Nutramax Labs), 20 to 40 mg PO q24h, as a glutathione source, because
decreased hepatic glutathione levels occur in hepatic lipidosis.
Response to Treatment and Prognosis
•
With aggressive nutritional and supportive care, approximately 60% to 85% of cats
with lipidosis respond within 3 to 6 weeks. Biochemical improvement (decreases in
bilirubin, ALP, and ALT) is usually seen within 1 to 2 weeks of initiating tube feeding.
Normalization may take several weeks. Do not remove the tube until the cat is eating
on its own for at least a week.
•
Recurrence is rare and there is no evidence of residual hepatic damage.
•
The earlier treatment is initiated, the better the prognosis.
Key Point
Monitor serum liver enzymes and institute nutritional support early in obese cats
that become inappetent or undergo rapid weight loss secondary to other disease processes.
•
Consider the potential for lipidosis in any obese cat placed on a reducing diet. Monitor
liver enzymes to evaluate for onset of lipidosis. Consider l-carnitine supplementation
(250 mg/cat/day) in obese cats undergoing dietary weight reduction.
CANINE VACUOLAR HEPATOPATHIES
The term vacuolar hepatopathy is used to describe the cytologic or histologic appearance
of hepatocytes, which contain either discrete cytoplasmic vacuoles (usually fat) or
cytoplasmic rarefaction, a term used to describe ballooned cells with less cytoplasmic
density but devoid of distinct vacuoles.
•
Cytoplasmic rarefaction is seen with either hepatocellular hydropic degeneration (increased
cellular water) or steroid hepatopathy (increased glycogen) and cannot be differentiated
without special stains. Glycogen is PAS positive.
•
Vacuoles containing lipid (fat) can be confirmed by oil red O staining of formalin-fixed,
non-paraffin embedded frozen tissue.
•
Vacuolar hepatopathy is a common histologic diagnosis in dogs. Clinical associations
are numerous and are listed in Table 71-7
.
Table 71-7
DIFFERENTIAL DIAGNOSES FOR CANINE VACUOLAR HEPATOPATHY
Glucocorticoid therapy
Including oral, parenteral, topical (eye, ear, skin)
Hyperadrenocorticism
Pituitary or adrenal origin
Atypical form (normal cortisol but increased adrenal steroid hormones, especially
17-OH progesterone)
Idiopathic vacuolar hepatopathy
Scottish terriers (progesterone?)
Others
Reactive hepatopathy
Common lesion in dogs with other systemic illnesses
Chronic (>4 weeks) illness (increased endogenous corticosteroids?)
Hepatic nodular regeneration
Hyperlipidemia
Diabetes mellitus
Idiopathic hyperlipidemia (miniature schnauzers, Shetland sheepdog, others?)
Hypothyroidism (severe)
Hepatocutaneous syndrome
Tetracycline administration
Glycogen and lysosomal storage disorders
Glucocorticoid Hepatopathy (Steroid Hepatopathy)
Glucocorticoid hepatopathy, or steroid hepatopathy, is a commonly recognized sequela
of glucocorticoid administration in dogs. Glucocorticoids cause hepatic glycogen accumulation
and hepatomegaly (see Table 71-4). Steroid hepatopathy is a benign, reversible hepatic
lesion that, with rare exceptions, is not associated with clinical liver dysfunction.
The most important clinical significance of this disorder is that it can easily be
mistaken for a more serious hepatic disease.
Key Point
To avoid unnecessary diagnostic and therapeutic measures, remember that increased
serum ALP activity and hepatomegaly are commonly caused by glucocorticoid therapy
in dogs.
•
Recognition of steroid hepatopathy often alerts the clinician to the presence of previously
unsuspected spontaneous hyperadrenocorticism in dogs without a history of glucocorticoid
therapy (see Chapter 33).
•
Cats are resistant to the hepatic effects of glucocorticoids, and development of steroid
hepatopathy is rare.
Etiology
•
Glucocorticoid hepatopathy has been associated with numerous glucocorticoids including
cortisone, prednisone, prednisolone, dexamethasone, and triamcinolone. Lesions of
steroid hepatopathy can develop within 7 to 14 days of corticosteroid administration.
•
Endogenous production of excess glucocorticoids caused by spontaneous hyperadrenocorticism
also results in steroid hepatopathy. Hepatic lesions are identical to those seen with
exogenous administration of glucocorticoids.
•
Dogs with atypical hyperadrenocorticism can also develop lesions of steroid hepatopathy.
These dogs present with clinical signs and laboratory find-ings typical for hyperadrenocorticism,
but cortisol levels in response to adrenocorticotropic hormone (ACTH) or after low-dose
dexamethasone are normal. Excess production of sex hormones, especially 17-hydroxyprogesterone,
has been described (see Chapter 33).
•
Individual variation in susceptibility to steroid hepatopathy also appears to play
a role.
•
Dogs that are chronically stressed (>4 weeks) due to other systemic illnesses (e.g.,
severe dental disease, chronic inflammation or infection, or neoplasia) may also develop
this hepatic lesion, presumably due to stress-induced endogenous glucocorticoid release.
Clinical Signs
•
Clinical signs reflect the systemic effects of hypercortisolism rather than hepatic
disease and include PU/PD and polyphagia in an otherwise healthy dog.
•
Dogs with other chronic illnesses do not typically have PU/PD but may show signs pertaining
to their underlying disorder.
Diagnosis
Suspect steroid hepatopathy in any dog with hepatomegaly and increased serum ALP activity
that has a history of recent glucocorticoid therapy and/or clinical signs of hyperadrenocorticism.
Key Point
Significant amounts of glucocorticoid can be absorbed from topical and ocular medications
as well as from oral and injectable preparations.
History and Physical Examination
•
Evaluate for previous glucocorticoid therapy within the past 3 months.
•
Hepatomegaly, which may be quite massive, frequently is detected.
•
Other findings in dogs with hyperadrenocorticism or exogenous glucocorticoid administration
include abdominal distention and thinning of the skin and haircoat.
Laboratory Evaluation
•
Increased serum ALP activity is the most consistent biochemical abnormality detected
in dogs with this hepatic lesion. After glucocorticoid administration, the initial
increase in ALP activity is attributed to the liver isoenzyme rather than the corticosteroid-induced
isoenzyme (CIALP; see previous discussion). This increase can occur within 2 to 3
days of glucocorticoid therapy and often is as high as 150 times normal. After 7 to
10 days the ALP elevation becomes progressively more attributable to increased CIALP.
The increase in CIALP can persist several months after exposure to corticosteroids.
•
Increased CIALP activity is a consistent finding in dogs with spontaneous hyperadrenocorticism,
and absence of this isoenzyme is uncommon in this disorder.
•
In contrast, ALT activity is normal or only mildly increased.
•
SBA concentrations are normal or mildly increased (<60μmol/L).
•
Total serum bilirubin, serum albumin, blood ammonia concentration, and hemostatic
tests typically are normal.
•
Other findings characteristic of hypercortisolism include mature neutrophilia, lymphopenia,
eosinopenia, monocytosis, and hypercholesterolemia.
Radiography and Ultrasonography
•
Hepatomegaly usually is detected on abdominal radiographs.
•
Ultrasonography reveals hepatomegaly and diffuse or multifocal increase in liver echogenicity.
Adrenomegaly may be detected in dogs with underlying spontaneous hyperadrenocorticism.
Liver Biopsy
•
Grossly, the liver is enlarged, smooth, pale, and friable. Microscopically, hepatic
lesions are characterized by cytoplasmic rarefaction in a patchy distribution.
•
PAS staining reveals that hepatocytes contain glycogen.
•
When hepatic biopsy suggests steroid hepatopathy and a history of glucocorticoid administration
is lacking, perform diagnostic tests for endogenous hyperadrenocorticism including
the atypical form (see Chapter 33). If tests for hyperadrenocorticism are negative,
evaluate for other disorders associated with vacuolar hepatopathy (Table 71-7).
Treatment
Key Point
Steroid hepatopathy does not require any specific therapy for the liver.
•
Steroid hepatopathy is reversible after withdrawal of exogenous glucocorticoids or
treatment of spontaneous hyperadrenocorticism.
•
The length of time required for complete resolution is unpredictable, varying from
weeks to months.
Vacuolar Hepatopathy in Scottish Terriers
Scottish terriers with diffuse vacuolar hepatopathy similar to steroid hepatopathy
have been recently described. Increased levels of adrenal steroids (especially progesterone
and 17-OH progesterone) have been documented. Progesterones, which are precursors
of glucocorticoids, have intrinsic glucocorticoid activity and can promote hepatic
glycogen accumulation.
Clinical Signs
Most dogs are asymptomatic although a few dogs may have mild PU/PD. Some dogs eventually
develop typical signs of spontaneous hyperadrenocorticism.
Diagnosis
•
Laboratory features include marked elevations in ALP activity (predominantly CIALP)
with normal GGT and ALT activity. Total bilirubin concentrations are normal. SBA concentrations
are normal or mildly increased.
•
On abdominal ultrasonography, the liver is enlarged and hyperechoic. Adrenal glands
appear normal. Liver biopsy reveals diffuse vacuolar hepatopathy, and PAS stains are
positive for glycogen.
•
Evaluation for spontaneous hyperadrenocorticism reveals normal ACTH stimulation and
low-dose dexamethasone suppression tests. However, testing for adrenal sex hormones
before and after ACTH reveals increased progesterone, 17-OH progesterone or other
adrenal steroid hormones with normal cortisol levels (Clinical Endocrinology Service,
College of Veterinary Medicine, University of Tennessee, 2407 River Drive, Room A105,
Knoxville, TN 37996; telephone: 865-974-5638).
Treatment
•
Although treatment with ketoconazole or mitotane will decrease ALP levels, no treatment
is recommended if dogs are asymptomatic. Preliminary results suggest this is a benign
disorder with a favorable long-term prognosis.
•
If clinical signs of hyperadrenocorticism develop, these dogs respond to standard
treatment (see Chapter 33).
Vacuolar Hepatopathy in Hyperlipidemic Miniature Schnauzers
Idiopathic hyperlipoproteinemia in miniature schnauzers is an inborn error of lipoprotein
metabolism characterized by fasting hypertriglyceridemia and hypercholesterolemia.
A partial decrease in lipoprotein lipase activity has been described in lipemic compared
with non-lipemic miniature schnauzers. Other breeds (such as Shetland sheepdogs) may
also be affected.
•
Affected dogs typically develop a marked vacuolar hepatopathy. Vacuoles contain both
fat and glycogen.
•
Rarely, they may develop a nodular liver with stromal collapse and clinical evidence
of hepatic insufficiency or severe sludging of bile and cholelithiasis.
Clinical Signs
Clinical findings in hyperlipidemic miniature schnauzers are referable to the lipemia
and not hepatic dysfunction.
•
Signs include lethargy, abdominal pain, decreased appetite, hepatomegaly, and seizures.
•
Recurrent acute pancreatitis is common and many dogs will eventually develop diabetes
mellitus.
Diagnosis
•
Laboratory findings include moderate to marked increases in ALP and GGT activity.
The corticosteroid-induced isoenzyme of ALP typically predominates. Increased ALT
activity is variable. SBAs are typically normal or mildly increased. Associations
with increased urine protein-to-creatinine ratio and microalbuminuria have also recently
been described.
•
Abdominal ultrasonography of the liver reveals hepatomegaly and diffuse increase in
echogenicity.
•
Cytology and biopsy are consistent with vacuolar hepatopathy.
Treatment
Manage hyperlipidemia with a low-fat, high-fiber diet and possibly lipid-lowering
drugs. General liver support with hepatoprotectants such as antioxidants and ursodiol
may be warranted (see Table 71-2). Avoid the use of corticosteroids.
Superficial Necrolytic Dermatitis (Hepatocutaneous Syndrome)
Superficial necrolytic dermatitis (SND), also called hepatocutaneous syndrome, necrolytic
migratory erythema, or metabolic epidermal necrosis, is a crusting, ulcerative dermatopathy
that appears to be a complication of hepatic or endocrine pancreatic disease in dogs
and cats.
Etiology
The etiology and pathogenesis of this syndrome are poorly understood. In dogs it is
most commonly associated with hepatopathy but has also been described with glucagon-producing
pancreatic endocrine tumors or glucagon-producing hepatic tumors. The syndrome has
also been reported in one cat with a pancreatic carcinoma and two cats with hepatopathy.
Many dogs with SND develop diabetes mellitus in the late stages of the disease.
The hepatopathy is usually of unknown origin, although liver disease associated with
anticonvulsant therapy and potential exposure to mycotoxins has been suggested. In
a recent report, chronic phenobarbital therapy was associated with SND in 44% of cases.
Biochemical evaluation, liver function testing, ultrasonographic appearance of the
liver, and hepatic biopsy indicate that the hepatopathy of dogs with phenobarbital-related
SND is distinctly different from the typical hepatic dysfunction and cirrhosis seen
in dogs with phenobarbital hepatotoxicity (see under “Phenobarbital-Associated Hepatic
Disease”).
Pathogenesis
The dermatologic lesions are similar to necrolytic migratory erythema in humans, which
is usually associated with hyperglucagonemia and hypoaminoacidemia secondary to a
glucagon-secreting pancreatic tumor. Hyperglucagonemia is believed to cause severe
hypoaminoacidemia by stimulating hepatic utilization of amino acids during gluconeogenesis,
resulting in a metabolic or nutritional imbalance affecting the skin. Intravenous
amino acid infusions can reverse the skin lesions in humans.
•
Plasma glucagon levels are increased and most plasma amino acids are decreased in
dogs with SND that have an underlying pancreatic endocrine tumor. Dermatologic signs
resolve if the tumor can be completely resected.
•
Plasma glucagon levels are reported to be normal or only mildly increased in dogs
with SND and hepatopathy. However, most of these dogs have severe hypoaminoacidemia
and some have responded to dietary supplementation with egg yolks, which are a rich
source of amino acids. The plasma amino acid levels in dogs with SND and hepatopathy
are significantly lower than in normal dogs or dogs with acute or chronic liver disease,
suggesting that the pathogenesis is not simply related to hepatic dysfunction. Hyperglucagonemia
may still play a central role in dogs with hepatopathy because glucagon exists in
numerous immunoreactive fractions and the assay for this hormone may be insensitive
to some glucagon species.
•
Alternatively, up-regulation of hepatic amino acid utilization in a hypercatabolic
state has also been hypothesized in dogs with SND and hepatopathy. Contributing factors
may include hormonal imbalances (glucocorticoids, thyroid supplementation, increased
adrenergic stimulation), phenobarbital therapy causing chronic hepatic microsomal
enzyme induction, and age-related changes in hepatocellular membrane stability (increased
hepatocyte responsiveness to adrenergic stimulation and enhanced amino acid utilization
for gluconeogenesis). Aging has also been associated with alterations in hepatic sensitivity
to glucagon.
•
Other nutritional deficiencies such as zinc, biotin, or essential fatty acids have
also been proposed.
Signalment and Clinical Signs
•
SND is a disease of older dogs (mean age of 10 years), especially males. Shetland
sheepdogs, West Highland white terriers, cocker spaniels, Scottish terriers may be
at increased risk.
•
Dogs are usually presented for dermatologic signs and lethargy and inappetence. Overt
signs of liver failure are unusual.
•
SND is characterized by bilaterally symmetrical crusting or erosive lesions of the
pads, mucocutaneous junctions, and pressure points (hocks and elbows). The dermatologic
manifestations are described in Chapter 49.
Diagnosis
Laboratory Evaluation
•
Abnormalities may include nonregenerative anemia, abnormal red cell morphology (poikilocytes
and target cells), mild to moderate increases in serum liver enzymes (ALP, ALT, and
AST), hyperglycemia, and mild to moderate increases in SBA concentrations. Hypoalbuminemia
and hyperbilirubinemia are less consistent findings.
•
Other potential features include hypoaminoacidemia, hyperglucagonemia, and elevated
insulin levels.
Ultrasonography
•
Ultrasound examination of the liver may identify a unique “honeycomb” or “Swiss cheese–like”
lesion consisting of 0.5- to 1.5-cm diameter hypoechoic regions surrounded by highly
echogenic borders. Dogs with SND related to glucagonoma do not have these characteristic
hepatic lesions on ultrasound.
•
The pancreas should be evaluated for nodules consistent with neoplasia, although most
glucagonomas in the pancreas have not been visualized on ultrasonographic examination.
Liver and Skin Biopsy
•
Dermatohistopathology is pathognomonic for SND (see Chapter 49). Lesions consist of
parakeratotic hyperkeratosis, intercellular or intracellular edema, and epidermal
hyperplasia. Bacteria, dermatophytes, and yeast can be secondary contaminants.
•
The liver is usually normal to increased in size and has a striking nodular appearance
that grossly mimics cirrhosis. However, the hepatic lesion is not true cirrhosis because
microscopically, the fibrous tissue is actually condensed stroma secondary to parenchymal
collapse and not due to increased collagen production. Areas of severe parenchymal
collapse contain hepatocytes with marked vacuolization (predominantly glycogen accumulation
but also some lipid deposition). Regions of collapse surround sharply demarcated nodules
of normal hepatic parenchyma. The hepatic lesion is most likely a reflection of underlying
nutritional, hormonal, or toxic abnormalities.
Treatment
If a glucagon-producing pancreatic tumor is diagnosed (only 10% of cases), it should
be surgically resected. When a toxic or metabolic cause of hepatic injury is identified
(e.g., phenobarbital therapy), there is the potential for resolution of the disorder
if the cause can be removed. However, most dogs with phenobarbital-associated SND
do not improve after discontinuing phenobarbital.
Nutritional Considerations
Key Point
The most effective treatment is IV administration of amino acids.
•
Give 10% amino acid solution (Aminosyn, Abbott Labs) at a dosage of 250 to 500 ml
per dog (or 25 ml/kg) slowly IV over an 8- to 12-hour period. Repeat this treatment
every 7 to 10 days as needed to control the dermatologic lesions. Some dogs show marked
improvement after the first treatment. If a patient has not responded after four treatments,
it probably won't.
•
Despite underlying evidence for hepatic disease, do not restrict protein intake unless
overt HE occurs. Feed a diet high in good-quality protein.
•
Consider oral protein supplementation with three to six egg yolks q24h or an amino
acid supplement (Pro-Mod, Ross Labs) 10 g per 7 kg of body weight (up to a maximum
of 40 g) q24h.
•
Give an oral multivitamin supplement (including vitamin E).
•
Give elemental zinc, 1 mg/kg PO q12h, for possible zinc deficiency.
•
Consider essential fatty acid supplementation.
Other Therapy
•
Refer to Chapter 49 for recommendations for treating the cutaneous lesions and secondary
cutaneous bacterial and fungal infections.
•
Manage diabetes mellitus with insulin therapy and a high-fiber diet.
•
Consider a long-acting somatostatin analogue (Octreotide, Sandostatin) to inhibit
glucagon release.
•
Parenteral corticosteroids are contraindicated because of the diabetic or prediabetic
state. Topical triamcinolone ointment q12h can be used on a short-term basis to decrease
pain and inflammation associated with deep fissures.
Prognosis
The prognosis is poor except in the small percentage of cases in which a glucagon-producing
pancreatic tumor is identified and removed. Most dogs die or are euthanized within
5 months of onset of the skin lesions.
HEPATIC AMYLOIDOSIS
Amyloidosis is a progressive systemic disease associated with extracellular deposition
of insoluble fibrillar proteins, which results in organ dysfunction. Amyloidosis in
dogs and cats is reactive and may occur secondary to chronic inflammatory, infectious,
or neoplastic disorders.
•
Amyloidosis is a familial disorder in the Chinese Shar-Pei dog and in Abyssinian cats.
Although concurrent amyloid deposition occurs in the liver, kidneys, spleen, and adrenal
glands, clinical manifestations of renal failure are most common.
•
Clinically significant hepatic involvement has been described in Chinese Shar-Pei
dogs and Siamese, Oriental shorthair, Devon Rex, Burmese, and domestic shorthaired
cats. Diffuse hepatic involvement predisposes patients to spontaneous hepatic rupture
because of hepatic vascular fragility and coexistent coagulopathy.
Signalment and Clinical Signs
•
Shar-Pei dogs and young cats are at increased risk.
•
Clinical signs include anorexia, PU/PD, vomiting, and jaundice.
•
Spontaneous rupture of the friable liver may cause acute hemoabdomen with signs of
lethargy, hypovolemic shock, or sudden death.
Diagnosis
Physical Examination
Physical examination reveals hepatomegaly. With spontaneous rupture, findings include
pale mucous membranes, hypothermia, and abdominal effusion.
Laboratory Evaluation
•
Potential laboratory findings include increased ALT activity, hyperbilirubinemia,
and increased SBAs.
•
Regenerative anemia may occur secondary to hepatic rupture and hemorrhage. Thrombocytopenia
and abnormally prolonged clotting times (which may be vitamin K responsive) have been
described in some cats.
•
Abdominocentesis reveals hemorrhagic abdominal effusion.
•
Concurrent renal amyloidosis may cause azotemia and proteinuria (see Chapter 77).
Radiography and Ultrasonography
•
Findings on abdominal radiographs may include hepatomegaly and possible abdominal
effusion.
•
Abdominal ultrasound may reveal a diffusely heterogenous liver with hypoechoic foci.
Liver Cytology and Biopsy
•
Diagnosis of hepatic amyloidosis requires liver biopsy with special stains (Congo
red) to confirm its presence. However, caution is advised since FNA for cytology or
liver biopsy can be complicated by severe hemorrhage.
•
Hepatic cytology may suggest amyloid based on the identification of pink amorphous
material adjacent to hepatocytes (with a modified Wright-Giemsa stain).
•
Grossly, the liver is pale, large, and friable with hemorrhages, hematomas, and capsular
tears.
•
Histologically, amyloid in the liver appears as a homogenous, amorphous, eosinophilic
material within the space of Disse and vessel walls.
Treatment
•
Use fluid therapy and blood transfusion to treat acute liver hemorrhage.
•
Give vitamin K1 for the coagulopathy (see Table 71-1).
•
Consider colchicine therapy (0.03 mg/kg PO q24–48h), which may be beneficial in Chinese
Shar-Pei dogs with hepatic amyloid.
•
The long-term prognosis in cats with hepatic amyloidosis is poor.
CANINE CHRONIC HEPATITIS
Chronic hepatitis is a heterogeneous group of necrotizing inflammatory diseases of
the liver. The clinical signs of chronic hepatitis initially are vague, such as anorexia,
weight loss, and depression; however, as hepatitis becomes advanced, signs of liver
failure develop, including jaundice, ascites, coagulopathy, or HE.
With few exceptions, the cause, pathogenesis, natural history, and optimal treatment
of these disorders in dogs is unknown. Because the laboratory and histopathologic
features often fail to determine the definitive etiology, combined clinical and histologic
criteria rather than etiologic classifications generally are used to categorize patients
with chronic hepatitis.
Idiopathic Chronic Hepatitis
Idiopathic chronic hepatitis is characterized by clinical signs and persistent laboratory
indicators of hepatic disease in association with chronic portal inflammation, piecemeal
hepatic necrosis, and fibrosis that frequently progresses to cirrhosis and liver failure.
Etiology
The disease must be considered idiopathic in most dogs; however, it is probable that
after an initial inciting hepatocyte injury, immune mechanisms are involved in perpetuating
the inflammation.
Signalment and Clinical Signs
•
The incidence of idiopathic chronic hepatitis appears to be highest in female dogs.
•
The mean age of onset is 5 to 6 years, but adult dogs of any age or breed can be affected.
•
Common signs include anorexia, depression, weakness, PU/PD, ascites, jaundice, weight
loss, and vomiting.
Diagnosis
The diagnosis is suggested by the clinical signs in conjunction with elevation of
serum liver enzyme activity. The diagnosis can be confirmed only by liver biopsy.
Historical and physical findings are consistent with chronic liver disease.
Laboratory Evaluation
•
Serum ALT activity usually is >10 times normal, reflecting ongoing hepatic injury
(inflammation). Serum ALP activity is usually >5 times normal, reflecting intrahepatic
cholestasis. Hyperbilirubinemia and bilirubinuria also are common.
•
Liver function tests such as SBA concentrations frequently are abnormal, reflecting
the degree of liver dysfunction.
•
Less consistent findings include hypoalbuminemia, hyperglobulinemia, mild nonregenerative
anemia, and abnormal hemostasis. Ascitic fluid, when present, typically is a transudate
or modified transudate.
Radiography and Ultrasonography
•
Radiographically the liver may appear small, and on ultrasonography nonspecific changes
in echogenicity may be detected.
Liver Biopsy
Liver biopsy and histopathology confirm the diagnosis.
•
The liver often is small and nodular because of the fibrosis and nodular regeneration
of cirrhosis.
•
The primary lesion is portal inflammation consisting primarily of lymphocytes and
plasma cells and occasional neutrophils and macrophages. The inflammation extends
into the hepatic lobule, causing piecemeal necrosis of hepatocytes. These lesions
are essential defining criteria for categorization as idiopathic chronic hepatitis.
•
Fibrosis usually is present.
Key Point
Perform a quantitative copper analysis and special stains for copper to determine
if increased hepatic copper content could be playing a role in chronic hepatitis.
Differential Diagnosis
When chronic hepatitis has been confirmed histologically, look for potential inciting
or perpetuating factors. Recognized types of chronic hepatitis are listed in Table
71-8
. If none are found, then idiopathic chronic hepatitis is the diagnosis.
Table 71-8
CANINE CHRONIC HEPATITIS
Familial predisposition
Bedlington terrier
Cocker spaniel (American and English)
Dalmatian
Doberman pinscher
Skye terrier
West Highland white terrier
Labrador retriever?
Infectious Hepatitis
Infectious canine hepatitis (experimental)
Acidophil cell hepatitis
Leptospirosis (serovars grippotyphosa and australis)
Drug- and Toxin-Induced Hepatitis
Anticonvulsants (phenobarbital, primidone)
Oxibendazole?
Carprofen?
Aflatoxin?
Lobular Dissecting Hepatitis
Idiopathic Chronic Hepatitis
Treatment
Treatment includes drug therapy, discussed below, and supportive measures, previously
described under “Principles of Treatment for Liver Disease” (see Table 71-1). Treatment
recommendations for idiopathic chronic hepatitis in dogs are empirical and clinical
studies are lacking.
Glucocorticoids
Glucocorticoid therapy is the cornerstone of treatment for canine chronic hepatitis,
yet efficacy has not been well documented. It is not clear whether immunosuppressive
doses (as used for immune-mediated disorders) or anti-inflammatory doses are optimal.
One retrospective study of dogs with chronic hepatitis suggested that immunosuppressive
doses were associated with prolonged survival. Anti-inflammatory doses may non-specifically
decrease hepatic inflammation and release of local cytokines, which contribute to
hepatic necrosis and fibrosis.
•
Consider glucocorticoid therapy when infectious diseases have been ruled out and characteristic
inflammatory infiltrates are detected on liver biopsy.
•
Give prednisolone, 1 to 2 mg/kg PO q24h, until clinical remission occurs; then taper
gradually to 0.5 mg/kg or the lowest effective dose for alternate-day maintenance.
•
Although prednisone must be converted by the liver to its active metabolite, prednisolone,
either drug can probably be used, based on the observation that humans with liver
disease can still rapidly convert prednisone to prednisolone.
•
Potential side effects of glucocorticoid therapy include sodium and water retention
(exacerbation of ascites), GI bleeding and catabolic effects (exacerbation of HE),
iatrogenic Cushing's disease, and secondary infections.
•
Monitor serum biochemistries every 1 to 2 weeks in the initial stages of treatment.
•
Consider a follow-up liver biopsy, 2 to 3 months after starting therapy, to confirm
remission of the disease.
Azathioprine
Azathioprine is an antimetabolite with anti-inflammatory and immune-modulating effects.
When prednisolone alone is ineffective or side effects become objectionable, consider
combination therapy using azathioprine and prednisolone (at a lower dose if side effects
are a problem).
•
Give azathioprine (Imuran, Burroughs Wellcome) at a dosage of 1 to 2 mg/kg PO q24h
for induction therapy. For maintenance, give the same dose once every other day while
giving prednisolone on the alternate days.
•
Because azathioprine may cause bone marrow suppression (neutropenia and thrombocytopenia),
monitor periodically with a CBC. Other less common side effects include GI toxicity,
pancreatitis, and hepatotoxicity.
Ursodiol
Ursodiol (Actigall) is a synthetic bile acid that has been useful in the treatment
of humans with chronic hepatitis. Its use in conjunction with anti-inflammatory therapy
in dogs with chronic hepatitis appears promising.
•
Ursodiol, a hydrophilic bile acid, is believed to be beneficial by expanding the bile
acid pool and displacing potentially hepatotoxic hydrophobic bile acids that may accumulate
in cholestasis. It also has membrane-stabilizing, cytoprotective, and immunomodulatory
effects on liver cells and promotes choleresis (see Table 71-2).
•
Ursodiol appears to be well tolerated in dogs when used at a dosage of 15 mg/kg PO
q24h.
Antioxidants
•
Free radicals may be generated in chronic hepatitis, and they may contribute to hepatic
injury. Antioxidants, such as vitamin E, are important to scavenge free radicals and
prevent oxidative injury.
•
Give vitamin E at a dosage of 50 to 400 IU PO q24h. Absorption of fat-soluble vitamins
may be decreased in chronic cholestatic hepatobiliary disorders. In this setting,
a water-soluble form of vitamin E may be preferable.
Prognosis
The response to treatment of idiopathic chronic hepatitis is variable, which is expected
because it probably represents a heterogeneous group of diseases.
•
Some dogs eventually can be taken off medication and remain in remission, but more
often therapy must be continued indefinitely.
•
Other dogs fail to respond, especially those that have advanced disease and cirrhosis
(the treatment of cirrhosis and its complications is discussed elsewhere in this chapter).
Hepatic Copper Accumulation and Chronic Hepatitis
Copper accumulation in the liver can be associated with significant hepatic injury
resulting in acute hepatitis, chronic hepatitis, and cirrhosis. It is one of the few
well-documented causes of chronic hepatitis in the dog. The severity of hepatic injury
is related to the amount of accumulated copper. Hepatic copper concentration in normal
dogs is less than 400μg/g (ppm) dry weight.
•
An inherited metabolic defect in biliary copper excretion causes chronic hepatitis
in Bedlington terriers. Copper concentrations range from 850 to 12,000μg/g dry weight
in affected Bedlingtons.
•
Hepatic damage in Bedlington terriers does not consistently occur until the copper
concentration exceeds 2000μg/g dry weight. The concentration at which abnormal hepatic
copper contributes to hepatic damage in other breeds is unknown and may vary among
breeds.
•
Copper accumulation in the liver may be a cause or an effect of chronic hepatitis.
Because copper normally is excreted in the bile, hepatic copper accumulation can also
occur secondary to any cholestatic hepatobiliary disorder (such as idiopathic chronic
hepatitis) that impairs bile flow.
•
Whether secondary copper accumulation can further contribute to hepatic injury is
unclear, but this is an important question with therapeutic implications.
•
Other breeds of dogs occasionally are diagnosed with chronic hepatitis and cirrhosis
accompanied by increased hepatic copper concentrations. At this advanced stage of
disease, it is difficult to know whether copper accumulation is a cause or an effect
of the chronic hepatitis. As a general rule, the higher the copper content, the more
likely it is to be a primary problem.
Copper-Associated Hepatitis in Bedlington Terriers
Bedlington terriers have a high incidence of an inherited (autosomal recessive) metabolic
defect in biliary copper excretion that leads to progressive intrahepatic copper accumulation
and chronic liver disease. Because of extensive inbreeding, the prevalence within
the breed is quite high (25% affected, 50% carriers).
Etiology
•
Hepatic copper content increases with age as a result of defective biliary copper
excretion.
•
Excess copper is bound to hepatic metallothionein and stored in hepatic lysosomes.
When hepatic copper accumulation is >2000μg/g dry weight, progressive hepatic injury
occurs, including focal hepatic necrosis, chronic hepatitis, and eventually cirrhosis.
This disease is similar but not identical to Wilson's disease in humans.
•
Hepatic injury associated with copper accumu-lation involves free radical damage to
hepatic mitochondria.
Clinical Signs
Clinical signs and presentation vary widely, depending on the stage of disease.
•
Most affected dogs are presented as young or middle-aged adults of either sex with
signs of hepatic failure of varying severity, including lethargy, depression, weight
loss, vomiting, and jaundice. Acute fulminant hepatic failure with rapid deterioration
and death occurs in rare instances.
•
Some middle-aged and older dogs are presented initially with end-stage liver disease
and cirrhosis. In these animals there is a more chronic, insidious clinical course
with similar but less severe signs. In the advanced stages of disease, cachexia, jaundice,
ascites, and HE can occur.
•
Affected dogs may be asymptomatic, especially young dogs in which copper is accumulating
but has not yet reached toxic hepatic concentrations.
Diagnosis
Suspect copper-associated hepatitis in any Bedlington terrier with historical, physical,
or biochemical evidence of hepatic disease or with vague, unexplained illness. Asymptomatic
dogs can be identified only by routine biochemical screening or liver biopsy. Definitive
diagnosis requires liver biopsy.
History
•
Acute, recurrent episodes of illness are common in many affected dogs. Stressful events
such as whelping, showing, shipping, or a change in environment can precipitate these
episodes.
•
Dogs initially presenting with end-stage cirrhosis often have no history of previous
episodes of hepatitis.
Physical Examination
•
Findings in dogs with acute hepatitis include depression, lethargy, and dehydration.
Hepatomegaly may occur. Jaundice may be detected within 48 hours of onset. Acute copper-induced
hemolytic anemia may be a contributing factor.
•
With advanced disease, dehydration, emaciation, ascites, HE, and jaundice may be detected.
The liver is small and not palpable.
•
Asymptomatic dogs are normal on physical examination.
Laboratory Evaluation
•
Biochemical findings vary with the stage of disease. Increased serum ALT activity
is the most sensitive laboratory indicator of this disease, although up to a third
of affected dogs will have normal ALT values. These are mostly younger dogs that are
in the early stages of the disease.
•
Other serum biochemical abnormalities typical of hepatic dysfunction eventually develop,
such as hyperbilirubinemia, bilirubinuria, hypoalbuminemia, increased SBA levels,
and prolonged PT and APTT.
•
Serum copper levels are not helpful for the diagnosis.
•
Acute release of copper from necrotic hepatocytes occasionally causes hemolytic anemia.
Laboratory findings include low packed cell volume (PCV), hemoglobinemia, and hemoglobinuria.
Radiography and Ultrasonography
•
Abdominal radiographs are unremarkable except when advanced stages of disease are
accompanied by microhepatica or ascites.
•
Ultrasonography of the liver may be normal in the early stages. As the disease progresses,
findings are indicative of diffuse liver disease or microhepatica and cirrhosis.
Liver Biopsy
Liver biopsy is indicated for definitive diagnosis and staging of the disease. Perform
liver biopsies in all Bedlingtons being considered for breeding. Dogs should be older
than 1 year because unaffected carrier dogs may have transient increases in hepatic
copper that return to normal by 1 year of age. The spectrum of gross and microscopic
features in the liver parallels the variable expression of the disease.
•
The liver can be grossly normal or swollen and smooth with accentuation of the lobules.
As cirrhosis develops, the liver decreases in size and there is a mixture of fine
and coarse nodules.
•
Histologically, H&E-stained hepatic tissue reveals dark granules in hepatocyte cytoplasm.
In the early stages, centrilobular hepatocytes are most affected, but later the distribution
is diffuse.
•
Histochemical stains for copper, such as rhodanine and rubeanic acid, are positive.
These stains correlate well with quantitative copper analysis when values exceed 850μg/g
dry weight.
•
Associated histologic hepatic damage is variable. In the most mildly affected animals,
only centrilobular copper granules are detected. This progresses to focal hepatitis,
lesions of chronic hepatitis, and eventually cirrhosis.
Hepatic Copper Analysis
Perform quantitative copper analysis on fresh hepatic tissue. Copper analyses are
available to veterinarians through the veterinary diagnostic laboratories of Michigan
State University and Colorado State University. Affected dogs have hepatic copper
concentrations of 850 to 12,000μg/g dry weight.
DNA Testing
•
DNA testing is available (www.vetgen.com) that identifies a linked marker located
in the chromosome close to the as-yet-unidentified causative gene.
•
Carrier, affected, or unaffected dogs can be characterized with 90% accuracy.
•
DNA-screened carriers or normal dogs should have annual liver enzyme evaluations.
Increases in liver enzyme activity should merit further evaluation of the liver.
Treatment
Specific measures to control hepatic copper accumulation are summarized in Table 71-9
. Base the choice of therapy for an individual patient on the severity of existing
hepatic damage.
Table 71-9
TREATMENT OF HEPATIC COPPER ACCUMULATION
Product
Formulation
Dosage
Side Effects
Comments
Chelate Systemic Cu
Penicillamine (Cuprimine, MSD; Depen, Wallace)
Cuprimine: 125- and 250-mg caps Depen: 250-mg tabs
15 mg/kg q12h PO given on an empty stomach to improve absorption; do not give concurrently
with any medication, including Zinca or vitamin and mineral supplements.
Anorexia and vomitingb are common; dermatologic drug eruption or autoimmune-like excretion.
Indicated when vesicular lesions of mucocutaneous junctions;c reversible renal disease.c
Causes systemic Cu chelation and urinary excretion. Indicated when hepatic Cu >1500
mg/g. Takes months to years to produce significant decrease in hepatic Cu concentration
(900 mg/g/year)d, but may produce subjective clinical improvement after a few weeks.
May have protective effect in liver beyond chelation: induces hepatic metallothionein,
which binds and sequesters Cu in non-toxic form? Not effective for treatment of Cu-associated
hemolysis.
Trientine dihydrochloridee (Syprine, MSD)
250-mg caps
15 mg/kg q12h PO. Give 1 hour before meals; do not give concurrently with any medication,
including zinca or vitamin/mineral supplement.
None noted as yet.
Use as an alternative to penicillamine if vomiting occurs. May be useful for treatment
of hemolysis by chelating Cu in blood. More expensive than penicillamine.
Decrease Intestinal Cu Absorption
Zinc acetate, sulfate, or gluconate to obtain therapeutic zinc
Many available
100-mg elemental zinc PO q12h for 2–3 months, then 50 mg PO q12h for maintenance.
Separate administration from meals by >1 hour.
Vomiting,f zinc-induced hemolysis at plasma levels >800 mg/dl.
Induces intestinal metallothionein, which preferentially binds Cu and decreases absorption.
Takes 3–6 months. Monitor plasma zinc every 2–3 months. Ideal level is 200–400 mg/dl.
Do not give concurrently with Cu chelators. Use when hepatic Cu is increased but is
<1500 mg/g dry weight.
Decrease Cu Intake
Cu-restricted diet
Prescription Diet l/d (Hill's) Homemade diets
Low Cu diet may slow further Cu accumulation but won't decopper the liver. Most commercial
diets have abundant Cu. Avoid mineral supplements, liver, shell fish, organ meats,
chocolate, nuts, mushrooms, cereals.
Antioxidant Therapy
g
Vitamin E
Many available
100–400 IU/day, PO.
None noted as yet.
Oxidative damage occurs in Cu-associated liver disease. Vitamin E protects the liver
against oxidant damage.
Cu, copper.
a
Penicillamine and trientine will chelate zinc (and decrease its absorption) when given
concurrently.
b
Often resolves after several weeks; start at a reduced dose and increase to maintenance
after a few days. Giving with food will decrease absorption, but a small amount of
milk, cheese, or bread given concurrently may decrease vomiting.
c
Rare complications; renal compromise more likely with Depen?
d
In Bedlington terriers; more rapid response in Doberman pinschers and other breeds.
e
Availability limited; may require a special order direct from manufacturer
f
Zinc acetate may be less irritating to the stomach than other formulations. To minimize
vomiting, open capsule and mix contents with small amount of tuna or hamburger.
g
Although vitamin C has antioxidant effects, it should be avoided in dogs with Cu accumulation
because it increases Cu's oxidative damage to the liver.
Key Point
Lifelong therapy is necessary because copper reaccumulates if treatment is stopped.
Management of the acute hepatic crisis also involves symptomatic and supportive care
to control electrolyte, acid-base, and fluid imbalances and HE (see Table 71-1). Treatment
of hemolytic anemia may require a blood transfusion.
Chelator Therapy
•
Treat affected dogs with copper accumulation (>1500μg/g dry weight) and chronic hepatitis
with a copper chelator such as penicillamine or trientine hydrochloride, which promotes
urinary copper excretion (see Table 71-9).
•
For treating an acute hemolytic crisis, trientine hydrochloride (but not penicillamine)
may be effective in chelating copper in the circulation.
•
There may be other protective effects of penicillamine besides depletion of hepatic
copper, because many Bedlington terriers on long-term therapy do not develop hepatic
failure despite continued elevated copper levels and ongoing hepatic damage. Penicillamine
may induce hepatic metallothionein, which binds and sequesters copper in a non-toxic
form. Additional effects that may be beneficial include anti-inflammatory, antifibrotic
activity, and immunomodulating effects.
Zinc Therapy
Use oral zinc therapy when hepatic copper concentrations are >400, but <1500μg/g dry
weight to prevent intestinal copper absorption and hepatic copper accumulation. Preliminary
information in Bedlington terriers and West Highland white terriers suggests it may
also deplete hepatic copper concentrations (see Table 71-9).
Dietary Therapy
Low copper diets are of little value once hepatic copper accumulation has occurred
(see Table 71-9) but may be helpful in the early stages to decrease intestinal copper
absorption.
Vitamin E Therapy
Administer vitamin E to all dogs with hepatic copper accumulation (see Table 71-9).
Hepatic injury associated with hepatic copper overload in Bedlington terriers appears
to involve free radical damage to hepatic mitochondria. Vitamin E therapy protects
against copper-induced hepatic damage experimentally.
Prognosis
•
Dogs with mild to moderate acute hepatic failure usually respond to supportive care.
•
If this disease is detected before severe hepatic failure occurs, many dogs can live
out their lives with penicillamine therapy.
•
The prognosis is poor if there is fulminant hepatic failure or chronic end-stage cirrhosis
and failure.
Prevention
•
Treatment of affected dogs with minimal hepatic injury is recommended in the hope
of preventing acute hepatitis or progression to cirrhosis.
•
Zinc therapy is a promising and less expensive alternative to penicillamine in this
setting.
•
Bedlington terriers used in breeding programs can be certified free of disease through
either the Canine Liver Registry at Purdue University or the Liver Registry of the
Orthopedic Foundation for Animals in Columbia, Missouri.
Chronic Hepatitis in West Highland White Terriers
West Highland white terriers (WHWTs) have increased risk for developing chronic hepatitis
and cirrhosis. Hepatic copper accumulation is found in many WHWTs with chronic hepatitis.
Hepatic copper accumulation in WHWTs is a familial trait, but the mode of inheritance
has not been established.
Etiology
The etiopathogenesis is unknown. The notable differences between the copper retention
in WHWTs and the disease in Bedlington terriers are that WHWTs do not accumulate copper
continuously throughout life, the peak hepatic copper concentration in WHWTs occurs
by 6 months of age and may even decrease after 1 year of age, and the magnitude of
the copper increase is generally lower in WHWTs than in Bedlingtons.
•
Many WHWTs have hepatic copper concentrations that range from 200 to 1500μg/g dry
weight, but rarely does the value exceed 2000μg/g dry weight and clinical illness
associated with excess hepatic copper accumulation is uncommon. Some dogs have high
hepatic copper (usually <3500μg/g dry weight) but no evidence of hepatic disease.
•
In WHWTs with chronic hepatitis, increased hepatic copper concentration (>2000μg/g
dry weight) appears to be a factor in some but not most affected dogs. When hepatic
copper concentrations are <2000μg/g dry weight, the copper may not be contributing
to hepatic injury. These dogs may have idiopathic chronic hepatitis.
•
Chronic hepatitis in WHWTs may also occur without increases in hepatic copper.
Clinical Signs
•
Affected animals in the early stages of copper accumulation or those with focal hepatitis
are usually asymptomatic.
•
When widespread necrosis occurs, nonspecific signs of liver disease include anorexia,
vomiting, diarrhea, lethargy, and jaundice.
•
With advanced disease, jaundice and ascites are common.
Diagnosis
Episodes of hepatic necrosis may be precipitated by stressful events such as whelping
or showing.
•
The earliest biochemical abnormality associated with hepatic necrosis is increased
ALT activity.
•
With advanced disease, laboratory findings include increased liver enzyme activity,
hyperbilirubinemia, increased SBA levels, hyperammonemia, and hypoalbuminemia. Copper-associated
hemolytic anemia has not been documented.
•
Liver biopsy for histopathology and quantitative copper analysis are required for
definitive diagnosis. Histologic features include copper granules (which are initially
centrilobular but become diffusely distributed with time), multifocal hepatitis, subacute
bridging necrosis, massive necrosis, and cirrhosis.
Treatment
If chronic hepatitis and cirrhosis are associated with increased hepatic copper (>1500μg/g
dry weight) in a WHWT, initiate treatment for hepatic copper accumulation as described
for Bedlingtons (see previous section) and in Table 71-9.
•
Preliminary evidence suggests that both penicillamine and zinc acetate can decrease
hepatic copper concentrations in WHWTs when given on a long-term basis (see Table
71-9).
•
Because hepatic copper accumulation is not continuous throughout life, mature dogs
with chronic hepatitis and hepatic copper concentrations <1500μg/g dry weight may
not require chelator therapy. Consider glucocorticoid therapy as described previously
for idiopathic chronic hepatitis.
Chronic Hepatitis in Doberman Pinschers
Doberman pinschers are at increased risk to develop chronic hepatitis and cirrhosis.
Hepatic copper concentrations are increased in most affected dogs.
Etiology
•
The underlying etiopathogenic mechanisms are unknown, but a genetic basis is suggested
by the high incidence in this breed. Middle-aged (5-7 years old) female dogs are at
increased risk.
•
Hepatic copper concentrations are increased (650–4000μg/g dry weight) in most affected
dogs but not to the same magnitude as seen with Bedlington terriers, suggesting a
different disease mechanism. The significance of the increased hepatic copper concentration
is still under debate.
•
Evaluation of Doberman pinschers with subclinical disease has recently provided further
information on the role of copper in this disorder. Screening of 106 clinically healthy
3-year-old Doberman pinschers in the Netherlands (for liver enzyme elevations, increased
SBA, or copper granules on FNA cytology of liver) with follow-up liver biopsy (including
quantitative copper) revealed subclinical hepatitis in 21% of the screened dogs. Affected
dogs were followed over 2 to 4 years with repeated liver biopsy and quantitative copper
analysis. Persistent hepatitis was only documented in dogs (5 females, 1 male) with
initial and final hepatic copper concentrations > 400μg/g dry weight (939 +/− 299μg/g
dry weight). Those dogs with repeatedly normal hepatic copper levels (<400μg/g dry
weight) did not have persistent hepatitis on liver biopsy (probably due to other reversible
causes). These findings suggest that there is a relationship among copper storage,
hepatocellular damage and hepatitis in Doberman pinschers.
•
An immune-mediated mechanism of disease in affected Dobermans is supported by the
finding of up-regulation of major histocompatibility complex class II antigens in
hepatocytes.
Signalment and Clinical Signs
•
Middle-aged females are predominantly affected.
•
Clinical signs may be mild or absent when the disease is fortuitously diagnosed in
the early stages. However, most dogs are diagnosed in the advanced stages of hepatic
failure.
•
Signs include anorexia, weight loss, lethargy, PU/PD, vomiting, diarrhea, ascites,
and jaundice.
•
Evidence of excessive bleeding (gingival bleeding, epistaxis, and melena) may be found.
•
Signs of HE often predominate in the terminal stages.
Diagnosis
Suspect chronic hepatitis in any Doberman pinscher (especially female) with clinical
and biochemical evidence of hepatic disease. Definitive diagnosis requires liver biopsy.
•
Common physical examination findings include ascites, jaundice, weight loss, and encephalopathy.
Splenomegaly (associated with portal hypertension) is common. The liver is small and
not palpable.
•
Laboratory findings frequently include increased ALP and ALT activity, hyperbilirubinemia,
bilirubinuria, hypoalbuminemia, increased SBA levels, and hyperammonemia. Coagulopathy
and thrombocytopenia are common in the advanced stages. Consider concurrent von Willebrand
disease in affected dogs with a bleeding disorder, because of its prevalence in this
breed (see Chapter 23).
•
Radiographic and ultrasonographic findings of microhepatica and ascites are consistent
with chronic liver disease and cirrhosis.
Liver Biopsy and Copper Analysis
•
Liver biopsy and histopathologic evaluation are necessary to confirm the diagnosis.
•
Grossly, the liver is small with micronodular or macronodular cirrhosis.
•
Histologically, the earliest lesion is inflammation and fibrosis around the small
hepatic vein branches. As the disease progresses, fibrous tissue septa radiate from
hepatic vein branches to the portal areas. In the late stage of the disease, lymphoplasmacytic
inflammation occurs around larger hepatic veins and portal tracts. Fibrosis and architectural
distortion consistent with cirrhosis are present.
•
Rhodanine and rubeanic acid stains usually are positive for copper, especially in
centrilobular regions. Stains for hepatic iron also are usually positive.
•
Quantitative copper analysis reveals mild to moderate copper accumulation (650-4000μg/g
dry weight).
Treatment
Optimal treatment has not been established.
•
Institute, as needed, symptomatic and supportive therapy for complications of hepatic
failure, including correction of fluid, electrolyte, and acid-base balance and treatment
of ascites, HE, and coagulopathies (see Table 71-1 and “Principles of Treatment for
Liver Disease”).
•
Therapy with anti-inflammatory or immunosuppressive drugs such as prednisolone with
or without azathioprine may be given, as described previously for idiopathic chronic
hepatitis. The efficacy of this treatment remains to be determined, but generally
the response is poor, possibly because most dogs are presented in advanced stages
of liver failure. Doberman pinschers with subclinical hepatitis treated with low doses
of prednisolone (0.1-0.5 mg/kg/day) did not show any significant improvement.
•
Consider penicillamine therapy, especially when subclinical hepatitis is diagnosed.
Five female Doberman pinschers with persistent subclinical hepatitis and increased
hepatic copper concentrations were treated with penicillamine (200 mg PO q12h for
4 months). Mean liver copper concentrations decreased from 1036 to 407μg/g dry weight
and improvement in liver histopathology was noted. Whether histopathologic improvement
was related to copper-chelating or anti-inflammatory effects of penicillamine could
not be ascertained.
•
Consider ursodiol therapy in this cholestatic disorder (see Table 71-2). Vitamin E
may also provide a beneficial antioxidant effect.
Prognosis
•
When Doberman hepatitis is diagnosed in the advanced stages, treatment usually is
unsuccessful. Most dogs die within weeks to months.
•
The prognosis may be more favorable if the disease is detected in the subclinical
stage, but the optimal therapeutic regimen remains to be determined.
Copper-Associated Hepatitis in Dalmatians
Chronic hepatitis and cirrhosis associated with hepatic copper accumulation has been
reported in dalmatians. Cholestasis is not a prominent biochemical or histologic feature
until later in the disease, suggesting that hepatic copper accumulation is more likely
to be caused by a familial metabolic disorder than to be secondary to altered hepatic
biliary copper excretion. Two of the dogs were related.
Clinical Signs
Most dogs presented initially with acute GI signs (anorexia, vomiting, diarrhea).
Diagnosis
•
Biochemical findings revealed markedly increased ALT activity (average 10.8 times
above normal range) and moderately increased ALP activity (average 5.5 times above
normal range).
•
Hyperbilirubinemia and hypoalbuminemia were seen with advanced disease. Glucosuria
(in the absence of hyperglycemia) and proteinuria were identified in some dogs.
•
Liver biopsy revealed hepatocellular degeneration, necrosis, and inflammation (predominantly
lymphocytes or neutrophils). The mean hepatic copper concentration was 3197μg/g dry
weight (range of 754–8390μg/g dry weight).
Treatment and Prognosis
•
Rapid progression of the disease was characteristic.
•
Copper chelator therapy may be beneficial if diagnosed before advanced liver disease
occurs (see Table 71-9).
Copper-Associated Hepatitis in Skye Terriers
Chronic hepatitis and cirrhosis associated with hepatic copper accumulation (range
of 358-2257μg/g dry weight) has been described in genetically related Skye terriers.
•
In the early stages, copper accumulation is absent, and biopsy findings indicate hepatocellular
degeneration with cholestasis and mild inflammation.
•
Chronic lesions are associated with intracanalicular cholestasis, chronic hepatitis,
and cirrhosis.
•
Skye terrier hepatitis is speculated to be a result of disturbed bile secretion with
secondary accumulation of copper.
Chronic Hepatitis in Cocker Spaniels
American and English cocker spaniels have an increased incidence of chronic hepatitis.
The cause is unknown. Hepatic copper accumulation does not appear to be a consistent
feature. Accumulation of alpha-1-antitrypsin in hepatocytes, a well-recognized cause
of cirrhosis in man, may be important in the pathogenesis.
Clinical Signs
•
Male cocker spaniels (average age 5 years) are at increased risk. Despite the chronicity
and severity of the underlying hepatic lesions, most affected dogs have a short duration
of clinical illness prior to presentation of usually 2 weeks or less.
•
Ascites is the most consistent presenting complaint. Other nonspecific signs of liver
disease include depression, mild jaundice, melena, dehydration, subcutaneous edema,
and coma.
Diagnosis
Key Point
Ascites and profound hypoalbuminemia (mean of 1.7 g/dl) are consistent findings.
•
Laboratory findings include mild anemia and mild to moderate increases in serum liver
enzyme activity (although some dogs have normal values).
•
The total serum bilirubin concentration is normal or mildly increased, suggesting
that cholestasis is not a key feature of the disorder. FSBA and PPSBA concentrations
are increased.
•
Ascitic fluid analysis is consistent with a transudate or modified transudate.
•
Radiographic findings include microhepatica and ascites. Ultrasonography often shows
a diffuse increase in echogenicity, although some dogs have a normal-appearing liver.
•
Liver biopsy reveals chronic periportal hepatitis (lymphocytes, plasma cells, and
lesser numbers of neutrophils), portal fibrosis, and micronodular or macronodular
cirrhosis.
•
Hepatic copper accumulation is not a consistent feature (200-550μg/g dry weight).
Treatment
Treatment for cocker spaniels with chronic hepatitis consists of general supportive
therapy for the complications of liver failure (see “Principles of Treatment for Liver
Disease” and Table 71-1).
•
Corticosteroid therapy may be beneficial.
•
The prognosis is poor and most dogs die within a month of diagnosis. Early diagnosis
appears to be the key to long-term survival.
Lobular Dissecting Hepatitis
Lobular dissecting hepatitis is a distinctive histologic form of chronic hepatitis
that occurs in young dogs. The median age of 21 affected dogs was 11 months, with
54% of dogs being 7 months or younger. It has been suggested that this is a specific
reaction pattern of the liver in neonatal and juvenile dogs to a wide variety of hepatic
insults. Standard poodles may be at increased risk for this form of chronic hepatitis.
Clinical Signs
Clinical features are those of advanced hepatic failure and portal hypertension. The
most consistent clinical finding is ascites.
Diagnosis
An attempt should be made to identify other causes of chronic hepatic disease (see
Table 71-8). Diagnosis requires liver biopsy.
•
Liver enzymes are typically increased.
•
Hypoalbuminemia and increased SBA concentrations are common.
•
The lesion is characterized histologically by lobular hepatitis; inflammation (lymphocytes,
plasma cells, macrophages, and neutrophils) is scattered throughout the hepatic lobule
rather than concentrated in periportal regions. Bands of collagen and reticulin fibers
dissect around single or small groups of hepatocytes and disrupt hepatic lobular architecture.
Copper stains are negative or moderately positive, consistent with secondary copper
accumulation. The liver is shrunken, pale to tan, with an almost smooth surface and
occasional hyperplastic nodules. Multiple acquired portosystemic shunts are present.
Treatment
Optimal treatment has not been determined, but general measures for management of
chronic liver failure are appropriate (see Table 71-1).
Acidophil Cell Hepatitis
Acidophil cell hepatitis has been described in Great Britain and is caused by an unidentified
transmissible agent that is probably viral but distinct from canine adenovirus type
1 (see Chapter 16).
•
It is characterized by acute or chronic hepatitis with slow progression to cirrhosis.
Acidophils, which are a consistent histologic feature of the disease, represent dying
hepatocytes.
•
Signs usually are typical of chronic liver failure.
•
Specific treatment has not been described, but general measures for management of
chronic liver failure are appropriate.
PHENOBARBITAL-ASSOCIATED HEPATIC DISEASE
Long-term phenobarbital therapy for control of seizures has been associated with chronic
hepatic disease and cirrhosis in dogs (see Table 71-4). Most dogs have been treated
with phenobarbital for months to years before the liver disease is apparent. Chronic
phenobarbital therapy also is rarely associated with SND (hepatocutaneous syndrome)
in dogs (see previous discussion in this chapter), which is distinct from the characteristic
phenobarbital-associated chronic hepatic disease and cirrhosis described in this section.
Etiology
•
The mechanism of phenobarbital-induced injury is not known, but higher doses, higher
blood levels (>40μg/ml), and long duration appear to be important factors.
•
Prior therapy with phenytoin or primidone may increase the risk of hepatotoxicity.
•
Hepatotoxicity has not been described in association with short-term injectable (IV
or IM) doses.
Clinical Signs
•
Clinical signs are those of chronic hepatic disease and include anorexia, lethargy,
weight loss, weakness, PU/PD, coagulopathy, and jaundice. Signs of overdosage (sedation
and ataxia) are consistent findings in dogs with phenobarbital hepatotoxicity. Ascites
and HE are most likely with advanced hepatic disease.
•
When impaired hepatic inactivation of phenobarbital causes increased blood levels,
seizure frequency may decrease.
•
An increased frequency of seizures may be related to the development of HE.
Diagnosis
Suspect phenobarbital-induced hepatopathy in any dog with a history of chronic phenobarbital
therapy and clinical and biochemical evidence of hepatic injury.
Laboratory Evaluation
Key Point
To detect early evidence of hepatic damage, routinely monitor liver enzymes (serum
ALP and ALT), total serum bilirubin, cholesterol, albumin, and phenobarbital levels
at least every 6 months in all dogs on chronic phenobarbital therapy.
•
Mild reversible increases in serum ALP and ALT activity (usually <5 times upper normal
limit) are common in dogs treated with phenobarbital related to microsomal enzyme
induction rather than hepatocyte injury. The increased ALP is usually attributable
to induction of the liver isoenzyme, but sometimes it results from increased CIALP.
•
Elevations in ALT and ALP activity greater than 5 times the upper limit of normal
or any elevation in AST may be an indicator of hepatotoxicity.
•
Increased SBA, hyperbilirubinemia, hypoalbuminemia, and hypocholesterolemia are better
indicators of significant hepatic damage.
Radiography and Ultrasonography
Key Point
Dogs on phenobarbital without hepatic injury often have an incidental finding of hepatomegaly.
With phenobarbital-induced chronic hepatic disease, radiographic findings may suggest
microhepatica due to cirrhosis. Ultrasonography is useful to further characterize
liver changes and to evaluate for other hepatic disorders.
Liver Biopsy
Perform a liver biopsy in dogs on phenobarbital when hepatic function tests are abnormal,
liver enzyme activities are greatly increased, clinical signs of hepatic dysfunction
are present, or ultrasonographic hepatic abnormalities are detected.
•
The most consistent histologic finding in dogs on phenobarbital therapy is hepatocellular
hypertrophy with a ground-glass appearance of the cytoplasm. Hypertrophy is due to
hyperplasia of smooth endoplasmic reticulum. This finding is commonly identified in
dogs without clinical or biochemical evidence of hepatic dysfunction and does not
warrant a change in drug therapy.
•
Chronic hepatic disease associated with phenobarbital therapy is characterized histologically
by biliary hyperplasia, nodular hyperplasia, fibrosis, and cirrhosis. A mild inflammatory
infiltrate (neutrophils, lymphocytes, plasma cells) is often present. These lesions
are by no means specific for phenobarbital-induced hepatic damage; however, in the
absence of other known causes of hepatic damage, circumstantial evidence would support
drug therapy as a likely cause.
Treatment
•
If possible, discontinue phenobarbital (gradual taper) in dogs with biochemical and
histologic evidence of hepatic disease (see Chapter 127).
•
Consider replacing phenobarbital with potassium bromide (15-30 mg/kg PO q12h with
food) as an anticonvulsant in dogs with phenobarbital-associated hepatotoxicity because
of its lack of hepatic metabolism or hepatotoxicity (see Chapter 127).
•
Clinical, biochemical, and histologic improvement can occur if phenobarbital is discontinued
or used at a reduced dosage prior to severe, end-stage liver disease. Clinical signs
improve within days to weeks of decreasing serum phenobarbital levels.
•
Additional supportive measures are important in managing dogs with phenobarbital-induced
hepatic disease. Control complications such as ascites and HE, as discussed previously
(see Table 71-1).
•
Consider hepatoprotectants such as ursodiol, SAMe, or vitamin E (see Table 71-2).
Corticosteroids are not indicated unless a significant inflammatory component is documented
histologically.
Prevention
Despite evidence for hepatotoxicity, phenobarbital is still the drug of choice for
long-term control of seizures in dogs.
Key Point
To decrease the likelihood of hepatotoxicity in dogs treated with phenobarbital, monitor
serum levels and adjust the dosage so that serum phenobarbital concentrations do not
exceed 35μg/ml.
HEPATIC CIRRHOSIS AND FIBROSIS
Cirrhosis is characterized by diffuse fibrosis and replacement of liver tissue with
structurally abnormal regenerative nodules.
Etiology
•
Cirrhosis is the irreversible end stage of chronic hepatic injury caused by infection,
hepatotoxins (e.g., copper or phenobarbital), immunologic injury (chronic hepatitis),
chronic cholestasis (chronic cholangitis in cats), or hypoxia. The common denominator
is hepatocyte death, which leads to repair by fibrosis and nodular regeneration.
•
When cirrhosis is fully developed, the histologic features of the original inciting
injury often are obscured by the cirrhotic changes.
Key Point
Fibrosis, regenerative nodule formation, and structural disruption further compromise
adjacent normal hepatocytes, intrahepatic blood flow, and intrahepatic bile flow;
thus, cirrhosis eventually reaches a point at which it is self-perpetuating.
Clinical Signs
Cirrhosis causes generalized hepatic dysfunction; thus, the clinical signs are those
of chronic hepatic failure. A combination of jaundice, ascites, and HE is highly suggestive
of cirrhosis.
Diagnosis
Laboratory Evaluation
Laboratory evidence of liver disease usually precedes the development of cirrhosis
but may go undetected because signs at that stage may be insidious and vague.
•
Serum liver enzymes usually are increased, although more modestly than during the
active injury stage of liver disease.
•
Circulating bilirubin, ammonia, and bile acids usually are increased, whereas serum
albumin usually is decreased. Hyperglobulinemia is sometimes seen.
•
Hemostatic abnormalities may reflect DIC, impaired hepatic synthesis of coagulation
factors, or vitamin K deficiency due to cholestasis (least likely).
Radiography and Ultrasonography
Key Point
Microhepatica is common in dogs with cirrhosis, whereas most cats with biliary cirrhosis
have hepatomegaly due to marked biliary hyperplasia.
Ultrasonography findings include microhepatica, irregular hepatic margins, focal lesions
representing regenerative nodules, and increased parenchymal echogenicity associated
with increased fibrous tissue. Splenomegaly and secondary acquired portosystemic shunts
also may be detected.
Liver Biopsy
Definitive diagnosis of cirrhosis requires liver biopsy.
•
Laparotomy or laparoscopy provides a better appreciation for the gross nodularity
of the liver than can be ascertained from blind percutaneous needle biopsy.
•
Microscopic features include fibrosis, regenerative nodules, and disruption of normal
hepatic architecture.
•
Concurrent inflammation may be detected, especially when the inciting cause of cirrhosis
is chronic inflammation.
Treatment
Because cirrhosis is essentially irreversible, treatment is mainly supportive, emphasizing
measures that control the various complications of severe generalized liver failure,
such as ascites, encephalopathy, gastric ulcers, coagulopathies, and infection (see
Table 71-1 and “Principles of Treatment for Liver Disease”).
If a probable cause or category of injury can be determined, specific treatment directed
at preventing further injury may slow progression of cirrhosis. If an underlying cause
can be determined (or is suspected), refer to the appropriate section of this chapter
for specific details regarding treatment. For example, adjust the drug regimen of
dogs receiving phenobarbital (see under “Phenobarbital-Associated Hepatic Disease”).
Penicillamine
Use a chelating agent such as penicillamine to treat dogs with copper-positive biopsies
(see Table 71-9). Penicillamine also has antifibrotic properties.
Anti-inflammatory Therapy
Treat dogs with histologic features of chronic hepatitis with anti-inflammatory drugs
(see under “Idiopathic Chronic Hepatitis”). Prednisolone has antifibrotic properties
as well as anti-inflammatory activity; however, weigh the benefits of corticosteroids
with the risks of potential adverse effects, such as GI ulceration and bleeding, increased
body catabolism that may exacerbate HE, and sodium retention that may exacerbate ascites
and edema.
Colchicine
Colchicine (0.025-0.03 mg/kg PO q24h) is an antifibrotic drug used to treat humans
with cirrhosis. It acts as a microtubule inhibitor, stimulant of collagenase activity,
and inhibitor of collagen deposition. Its benefit in dogs with cirrhosis is unproven.
The major side effects in dogs are nausea, vomiting, and diarrhea. In humans, other
side effects include bone marrow toxicity and myoneuropathy.
CONGENITAL PORTOSYSTEMIC SHUNTS
Portosystemic shunts (PSSs) are vascular communications between the portal and the
systemic venous systems that allow portal blood to access the systemic circulation
without first passing through the liver. Clinical signs of HE result from inadequate
hepatic clearance of enterically derived toxins such as ammonia, mercaptans, short-chain
fatty acids, GABA, and endogenous benzodiazepines. Decreased hepatic blood flow and
lack of hepatotrophic factors result in hepatic atrophy.
Etiology
PSS in dogs and cats can be a congenital malformation or develop secondary to portal
hypertension.
Single Congenital Portosystemic Shunts
Single PSSs are most common and occur as a congenital developmental malformation.
Single shunts are not associated with portal hypertension. Single PSSs can be further
categorized as intrahepatic or extrahepatic.
Single Intrahepatic Shunts
•
Single intrahepatic shunts provide a communication between the portal vein and the
caudal vena cava, often via the left hepatic vein. This results from failure of the
fetal ductus venosus to close.
•
This type of shunt is most frequent in large-breed dogs.
Single Extrahepatic Shunts
•
Single extrahepatic shunts usually connect the portal vein or one of its tributaries
(left gastric or splenic vein) with the caudal vena cava cranial to the phrenicoabdominal
veins. Less frequently, the anomalous vessel will enter the azygous vein.
•
This type of shunt is most frequent in small-breed dogs and cats.
Multiple Extrahepatic Portosystemic Shunts
Multiple extrahepatic PSSs are collateral vessels that develop as a compensatory response
to sustained portal hypertension. These shunts are rudimentary, nonfunctional, microvascular
communications between the portal and the systemic veins that are present in normal
dogs and cats. With sustained portal hypertension, these vessels enlarge and function
to shunt blood into the lower-pressure systemic circulation, thus decreasing portal
pressure.
•
Multiple extrahepatic PSSs occur secondary to disorders causing portal hypertension
(Table 71-10
).
Table 71-10
DISORDERS ASSOCIATED WITH PORTAL HYPERTENSION AND MULTIPLE EXTRAHEPATIC PORTOSYSTEMIC
SHUNTS
Chronic hepatitis and cirrhosis
Lobular dissecting hepatitis
Congenital hepatic arteriovenous fistula
Portal vein hypoplasia, including hepatoportal fibrosis variant
Postoperative complication of congenital portosystemic shunt ligation
Chronic biliary obstruction
Portal vein obstruction (thrombosis, neoplasia, extraluminal compression)
Caudal vena cava or main hepatic vein obstruction (kinking, thrombosis, neoplasia)
•
These shunts usually appear as a tortuous plexus of vessels that communicate with
the caudal vena cava in the area of the kidneys.
•
Diagnosis and therapy are directed toward the underlying liver or portal vascular
disorder.
Clinical Signs
The following discussion focuses on single congenital PSS. Clinical signs of congenital
PSS are usually referable to the CNS, GI system, or urinary tract (see also under
“Signalment,” “History,” and “Physical Examination” in this section).
Central Nervous System Signs
•
Signs of HE often predominate, including episodic weakness, ataxia, head pressing,
disorientation, circling, pacing, behavioral changes, amaurotic blindness, seizures,
and coma.
•
Hypersalivation, seizures, and blindness are more common in cats with PSS than in
dogs.
•
Clinical signs of encephalopathy tend to wax and wane and are often interspersed with
normal periods, reflecting the variable production and absorption of neurotoxic enteric
products.
Gastrointestinal Signs
•
GI signs of intermittent anorexia, vomiting, and diarrhea are common and are not necessarily
accompanied by overt signs of HE.
•
Stunted growth, weight loss, failure to gain weight, and unthriftiness are common.
Urinary Signs
Key Point
Urate urolithiasis is an important complication of PSS because of increased urinary
excretion of ammonia and uric acid. Renal, cystic, and urethral calculi usually are
green and contain an ammonia or uric acid component.
•
If urolithiasis is a complicating feature, pollakiuria, dysuria, and hematuria may
occur.
•
Psychogenic polydipsia and subsequent polyuria are frequent findings in dogs.
Diagnosis
Suspect congenital PSS in the following patients:
•
Young dogs and cats with intermittent CNS, GI, or urinary tract signs
•
Young animals with unexplained weight loss, failure to grow, unthriftiness, or hypoglycemia
•
Dogs (except dalmatians and bulldogs) or cats with urate urolithiasis
•
Dogs and cats of any age with clinical and biochemical evidence of hepatic insufficiency
(especially HE) and absence of histologic evidence of severe intrahepatic disease
Key Point
In young animals with clinical features of a congenital PSS but without a demonstrable
shunt on portography or portal scintigraphy, consider hepatic microvascular dysplasia
(see later in this chapter).
Although congenital PSS may be suspected because of historical, physical, laboratory,
and radiographic findings, a definitive diagnosis requires identification of a shunt
by ultrasonography, contrast radiography, transcolonic or transplenic portal scintigraphy,
or exploratory laparotomy.
Signalment
•
Congenital PSS is more common in purebred than in mixed-breed dogs. The genetic basis
is unknown, although an increased incidence has been recognized in Yorkshire terriers,
miniature schnauzers, Irish wolfhounds, Cairn terriers, Maltese dogs, Australian cattle
dogs, Old English sheepdogs, Labrador retrievers, and golden retrievers.
•
Domestic shorthaired cats are affected more commonly than are purebred cats. Of affected
purebreds, Persian and Himalayan cats are at increased risk.
•
No sex predilection has been noted. Affected male dogs and cats are often cryptorchid.
•
Age is an important diagnostic clue, because most animals develop signs by 6 months
of age. A congenital PSS is also a diagnostic consideration in middle-aged and older
dogs, because signs may be subtle and occasionally animals go undiagnosed until as
old as 10 or 12 years of age.
History
•
Many affected animals have a history of stunted growth or failure to gain weight compared
with unaffected littermates.
•
Prolonged recovery after general anesthesia or excessive sedation after treatment
with tranquilizers, anticonvulsants, or organophosphates can be attributed to impaired
hepatic metabolism of these substances.
•
Signs of encephalopathy may be exacerbated by a protein-rich meal; GI bleeding associated
with parasite infection, ulcers, or drug therapy; and administration of methionine-containing
urinary acidifiers or lipotrophic agents.
•
Clinical improvement after fluid therapy is common and most likely attributed to correction
of dehydration and promotion of urinary excretion of ammonia and other toxins. Improvement
with broad-spectrum antibiotic therapy reflects the effect of antibiotics on the toxin-producing
intestinal flora.
Physical Examination
•
Findings may be unremarkable except for small body stature or weight loss.
•
The neurologic examination is normal or, if overt signs of HE are present, neurologic
findings are consistent with diffuse cerebral disease.
•
Ascites and edema are rare unless the shunt is complicated by marked hypoalbuminemia
(less than 1 g/dl).
•
Many affected cats have copper-colored irises.
Laboratory Evaluation
Routine hematologic and biochemical findings often are unremarkable. Although individual
parameters might be only mildly abnormal, test results often reflect a pattern suggesting
hepatocellular dysfunction in the absence of significant cholestasis or hepatocellular
necrosis.
•
Hematologic findings include microcytosis, target cells, poikilocytosis (especially
in cats), and mild non-regenerative anemia. Microcytosis is associated with abnormal
iron metabolism (impaired iron transport or iron sequestration) rather than absolute
iron deficiency. These RBC changes can be subtle but important diagnostic clues in
an otherwise normal CBC.
•
Urinalysis findings include dilute urine, ammonium biurate crystalluria, and mild
bilirubinuria.
•
Coagulation tests are normal.
•
Hepatocellular dysfunction is suggested by hypoproteinemia, hypoalbuminemia, hypoglobulinemia,
hypoglycemia, decreased BUN, and mild hypocholesterolemia. Hypoalbuminemia is a consistent
finding in dogs but not in cats. Total serum bilirubin concentration is normal.
•
Serum liver enzyme (ALP and ALT) levels are normal to mildly (2-4 times) increased,
consistent with a lesion of hepatic atrophy and minimal hepatocellular injury or intrahepatic
cholestasis. Increases in ALP activity in these young animals may actually be due
to the bone isoenzyme.
•
Measure SBA concentrations to document hepatic dysfunction in dogs and cats suspected
of having congenital PSS. Fasting SBA concentrations may be normal or increased, but
PPSBA concentrations are consistently abnormal and usually exceed 10μmol/L.
Key Point
The pattern of a normal FSBA concentration with a markedly increased PPSBA level is
characteristic of PSS. A consistently normal PPSBA concentration makes a diagnosis
of congenital PSS highly unlikely.
•
In preliminary studies in dogs with congenital PSS, the sensitivity of urinary bile
acids (UBA) (urine sample taken 4 to 8 hours after eating) was 100%, compared with
84% for FSBA and 98% for PPSBA.
•
Hyperammonemia is a common finding in animals with PSS, but fasting blood ammonia
concentration may be normal. The ATT is consistently abnormal and is equal in sensitivity
to PPSBA concentrations for detecting hepatic dysfunction associated with PSS (see
“Diagnostic Strategy for Liver Disease”).
Radiography and Ultrasonography
Stabilize the patient by instituting therapy for HE prior to giving anesthesia for
portography or for surgical correction (see under “Medical Therapy” in this section).
•
Abdominal radiography
commonly reveals microhepatica in dogs but not in cats. Mild renomegaly of unknown
clinical significance also is common. Intra-abdominal detail may be poor because of
lack of abdominal fat. Ammonium urate urinary calculi may be visible on survey radiographs
if they contain substantial amounts of magnesium and phosphate.
•
Routine abdominal ultrasonography
may demonstrate intrahepatic and extrahepatic shunts. Intrahepatic shunts are more
reliably detected with this procedure than are extrahepatic PSS. Urinary calculi also
can be identified.
•
Transcolonic portal scintigraphy
using technetium 99m pertechnetate is a non-invasive and highly sensitive test (available
at some referral institutions) for detecting whether a shunt is present; however,
it does not provide reliable anatomic information such as the type and location of
the shunt.
•
Ultrasound-guided transplenic portal scintigraphy
has recently been described. This procedure provides more anatomic detail with less
radiation exposure than transcolonic portal scintigraphy.
•
Positive-contrast portography
is the procedure of choice to accurately characterize the type and location of a PSS.
Techniques include splenoportography, mesenteric (or jejunal) portography, and cranial
mesenteric or celiac arterial portography. An operative mesenteric portogram is preferred
because it allows evaluation of the entire portal vein, does not require special equipment,
and results in few complications.
Technique for Mesenteric Portography
1.
Place the animal under general anesthesia (see Chapter 2 for anesthesia of the patient
with liver disease).
2.
Isolate a loop of jejunum through a ventral midline incision.
3.
Place two ligatures around a jejunal vein, and place an over-the-needle catheter (Abbocath,
Abbott) within the vessel. Tie the ligatures and secure the catheter to the vessel.
4.
Temporarily close the abdominal incision.
5.
Inject a water-soluble contrast agent (Conray, Mallinckrodt, or Iohexol, Winthrop)
as a bolus (2 ml/kg) into the catheter.
6.
If a rapid film changer is not available, take a lateral and ventrodorsal radiograph
as the final milliliter is injected.
Interpretation
•
If a single PSS is identified, it should be further characterized as intrahepatic
or extrahepatic, because this has important surgical ramifications (see Chapter 72).
•
If multiple extrahepatic PSSs are identified, portal pressure determination and gross
and microscopic findings of the liver are used to distinguish between congenital and
acquired causes (see the next section).
•
Failure to visualize the intrahepatic portal system is not a reliable indicator of
vascular atresia but may correlate with a greater occurrence of postoperative complications
after complete or partial shunt ligation.
Liver Biopsy
•
The liver is grossly small but otherwise fairly normal in appearance.
•
In some animals, biopsy findings are unremarkable.
•
Liver biopsy lesions can be subtle but most consistently reveal hepatocyte atrophy
with small or absent portal veins. Varying degrees of arteriolar hyperplasia, lipogranulomas,
and biliary hyperplasia may be seen. Hepatocellular vacuolization is sometimes noted
and may be severe. These biopsy findings reflect decreased portal blood flow and are
indistinguishable from those seen in animals with hepatic microvascular dysplasia,
primary hypoplasia of the portal veins, or portal vein obstruction or after experimental
surgical creation of a PSS.
•
Microscopic CNS abnormalities include polymicrocavitation of the brain stem and cerebellum
and an increased number of astrocytes in the cerebral cortex.
Treatment
Surgery
•
The treatment of choice for dogs and cats with a single PSS is surgical attenuation
or complete ligation of the shunt (see Chapter 72). Although complete shunt ligation
is preferred for improved long-term outcome, concurrent hypoplasia of the portal system
(whether as a primary vascular disorder or secondary to prolonged portosystemic shunting),
may increase intrahepatic vascular resistance and predispose the patient to portal
hypertension. Consequently, attenuate the shunt to the maximum degree that can be
tolerated without causing portal hypertension.
•
Highly successful alternative approaches for gradual progressive closure of the shunt
use surgically placed ameroid constrictors or cellophane bands around the shunt or
intravascular thrombogenic coils within the lumen of the shunt (see Chapter 72). Theoretical
advantages of slow attenuation of the shunt (over days to weeks) include reduced risk
of postoperative portal hypertension and neurologic dysfunction and decreased surgical
and anesthetic time.
Key Point
Gradual occlusion of the shunt should allow time for hepatic regeneration, expansion
of the portal vascular system, and accommodation of portal blood flow without portal
hypertension.
Medical Therapy
Medical management of dogs and cats with PSS is palliative and is directed primarily
at control of HE with a moderately protein-restricted diet supplemented with soluble
fiber, lactulose, and neomycin (see Table 71-1).
•
The short-term response to therapy for HE is often dramatic, and the animal usually
is clinically normal even prior to surgical shunt ligation.
•
If surgical shunt correction is not feasible or is declined by the owner, long-term
medical management may control clinical signs for as long as 2 to 3 years. However,
medical management of PSS does not reverse the progressive hepatic atrophy and alterations
in carbohydrate, lipid, and protein metabolism.
•
Acute decompensation of encephalopathy requires fluid therapy for correction of dehydration,
correction of electrolyte and acid-base imbalances, and maintenance of blood glucose
levels (see Table 71-1).
•
When severe CNS depression or coma prevents the oral administration of lactulose and
neomycin, administer these drugs via an enema (see Table 71-1).
•
Identify and correct precipitating causes of ence-phalopathy whenever possible, such
as hypogly-cemia, GI bleeding from hookworm infection, and hypokalemia (see Table
71-1).
•
Management of urate urolithiasis is discussed in Chapter 79.
Perioperative Complications
Other perioperative complications of shunt ligation include seizures, portal hypertension,
intra-operative hypothermia and hypoglycemia, anesthetic complications, fever and
positive blood cultures, portal vein thrombosis, coagulopathy, acute pancreatitis,
and cardiac arrhythmias.
Postoperative Seizures
Occasionally, seizures or status epilepticus are a complication of surgical shunt
ligation. Dogs older than 18 months of age may be at increased risk. The pathogenesis
is obscure, but seizures do not appear to be caused by simple hypoglycemia or HE.
It is possible that the brain may have adapted to an altered metabolism. The prognosis
for recovery from this complication is poor. Long-term anticonvulsant therapy is often
required if the patient survives the acute postoperative period.
Key Point
Evaluate for identifiable metabolic causes of postoperative seizures, such as hyperammonemia,
hypoglycemia, hypoxia, electrolyte imbalances, acid-base imbalances, and systemic
hypertension.
•
In addition to routine management of HE and correction of underlying metabolic imbalances
(including thiamine administration in cats), manage seizures with IV phenobarbital
(at reduced doses) or loading doses of potassium bromide (see Table 71-1 and Chapter
127).
•
If seizures cannot be controlled, administer IV propofol to induce general anesthesia
for 12 to 24 hours. Place an endotracheal tube and use a respirator to maintain pO2
and pCO2. Maintain anesthesia by propofol drip or isoflurane gas anesthesia. Consider
mannitol (0.5-1 g/kg IV) for control of cerebral edema (see Table 71-1).
•
Consider preoperative anticonvulsant therapy to help control or prevent postoperative
seizures in PSS patients (see Table 71-1). Felbamate, levetiracetam, or topiramate
are possible choices in dogs. Because of the short half-life of these drugs, therapeutic
blood levels can be reached within a week. Topiramate can be used in cats. If postoperative
seizures do not occur, anticonvulsant therapy can be tapered over a 4-week period
after surgery and then discontinued.
Prognosis
Dogs
The prognosis for resolution of signs after total surgical ligation of the shunt in
dogs is excellent if the dog survives the immediate postoperative period.
•
In dogs with partial shunt ligation, the prognosis is not as good. Although clinical
signs may resolve after surgery, and response appears favorable in the first few years,
long-term follow-up (>3 years) suggests recurrence of signs will occur in 40% to 50%
of dogs with partial shunt ligations.
•
If clinical signs recur in dogs that have had partial ligation, reevaluate by transcolonic
portal scintigraphy. If shunting persists, perform surgical exploration for complete
shunt closure by suture ligation or ameroid constrictor.
Cats
The response to surgical correction of a congenital PSS in cats appears to be less
encouraging than in dogs. With partial shunt ligation, clinical improvement is usually
noted after surgery, but relapse of neurologic abnormalities is common. Persistent
seizures and blindness are also more likely to occur when partial rather than total
ligation is performed.
HEPATIC MICROVASCULAR DYSPLASIA
Hepatic microvascular dysplasia (HMD) refers to congenital histologic vascular abnormalities
of the liver in dogs (and rarely in cats) that result in abnormally increased SBA
concentrations and may be associated with clinical signs of portosystemic shunting
of blood. It has been hypothesized (but not proven) that HMD results in intrahepatic
microscopic portosystemic shunting of blood. Because the shunt fraction is small compared
with shunting that occurs with a single macroscopic congenital PSS, the clinical signs
are less severe (or absent) and SBA concentrations are only mildly increased. Portal
hypertension is not a clinical feature of HMD for most affected dogs.
•
The relationship among HMD, congenital PSS and primary hypoplasia of the portal veins
is unclear. These disorders have similar hepatic histologic features (hepatic arteriolar
hyperplasia, small or absent portal veins, and hepatic lipogranulomas), which are
a stereotypical histologic response of the liver to inadequate portal vein flow. These
same histologic findings also develop after experimental surgical creation of a PSS.
These disorders may represent varying expressions of a more general developmental
vascular disorder.
•
It is likely that most animals with PSS have some degree of HMD, which may explain
the persistence of increased SBAs and histologic vascular lesions following complete
surgical ligation of a PSS in some cases. If vascular dysplasia accompanying PSS is
severe enough (e.g., primary hypoplasia of the portal veins), complete PSS ligation
cannot be performed without the development of portal hypertension.
Etiology
HMD has been studied extensively in Cairn terriers, where it is believed to be an
inherited disorder. A polygenic mechanism of inheritance is suspected.
Clinical Signs
Affected dogs and cats may be asymptomatic (especially true for Cairn terriers) or
show signs similar to those seen with congenital PSS.
•
Drug intolerance for products dependent on hepatic metabolism may be the only manifestation
in otherwise asymptomatic patients.
•
In symptomatic animals, signs are variable and include anorexia, lethargy, vomiting,
diarrhea, PU/PD, dysuria and hematuria (due to urate urolithiasis), and HE and seizures.
Diagnosis
Consider HMD in dogs and cats with clinical features of congenital PSS, increased
SBA concentrations, and typical liver biopsy findings and in those that do not have
a demonstrable shunt.
•
In symptomatic animals, it is essential to rigorously pursue the diagnosis of congenital
PSS (as described in the previous section), because the clinical and histologic features
of HMD alone are similar to those of congenital PSS. Specific surgical correction
is the optimal treatment if a shunt is identified.
•
Also consider the possibility that a dog or cat with increased SBA concentrations
may have HMD and be asymptomatic for the disorder (especially Cairn terriers). An
animal with HMD may have clinical signs due to an unrelated non-hepatic disease. Detection
of increased SBA concentrations may focus diagnostic efforts on the liver, causing
the clinician to overlook the true cause of the clinical signs.
Signalment
•
Yorkshire terriers and Cairn terriers are most commonly affected. However, HMD has
been diagnosed in many other small breeds of dogs, such as Maltese, dachshund, poodle,
Shih Tzu, Lhasa apso, cocker spaniel, and WHWTs.
•
Domestic shorthair cats are at increased risk for HMD.
•
Dogs with HMD tend to be older at presentation than dogs with congenital PSS.
Physical Examination
Physical examination is often within normal limits unless signs of HE are present.
Laboratory Evaluation
•
Dogs with HMD consistently have increased SBA concentrations, but not as increased
as in dogs with PSS. A shunting pattern is typically seen: normal or low FSBA concentrations
with moderately increased PPSBA concentrations. Indocyanine green clearance values
are also consistently abnormal.
•
Biochemical findings in dogs with HMD are usually unremarkable except for mild to
moderate increases in ALT activity.
Key Point
As opposed to dogs with congenital PSS, dogs with HMD do not usually have microcytosis,
hypoalbuminemia, decreased BUN, hypocholesterolemia, hypoglycemia, hyperammonemia,
or ammonium biurate crystalluria.
•
Preliminary results show that decreased protein C concentrations occur in 98% of dogs
with congenital PSS but only 30% of dogs with HMD. Whether this test will be useful
for differentiation of PSS versus HMD awaits further clinical studies.
Radiography and Ultrasonography
•
Radiographically, the liver is usually normal in size but may be equivocally small
in some cases.
•
On ultrasonography the liver may be subjectively decreased in size and the portal
vasculature may be decreased. Bladder or kidney stones are uncommon.
•
Transcolonic portal scintigraphy is usually normal or only mildly abnormal in HMD,
as opposed to the increased shunt fractions seen with congenital PSS. Since this is
a relatively sensitive method for detecting congenital PSS, a normal exam would make
PSS unlikely and support a diagnosis of HMD.
•
Portography will fail to identify a large shunting vessel. Cairn terriers with HMD
have abnormal truncation of the terminal branches of the portal veins and delayed
clearing of contrast, which gives the parenchyma a “blush” appearance.
Liver Biopsy
A wedge biopsy of the liver is preferred over a needle biopsy because the vascular
lesions are subtle and a wedge biopsy provides more hepatic lobules for evaluation.
•
Grossly, the liver is normal in size and color compared with the small liver seen
with congenital PSS.
•
The histologic features of HMD are typical for inadequate portal vein flow and include
small or absent portal veins, hepatic arteriolar hyperplasia, and hepatic lipogranulomas.
These findings are similar to those seen in dogs with congenital PSS, primary hypoplasia
of the portal vein, portal vein obstruction, or after experimental surgical creation
of a PSS.
Key Point
Biopsy more than one lobe in HMD because the histologic lesions can vary among liver
lobes, with some lobes appearing very abnormal and others appearing normal.
Treatment and Prognosis
•
No treatment is indicated for animals whose symptoms are subclinical.
•
If clinical signs of HE are present, they can often be successfully managed with a
moderately protein-restricted diet. Additional therapy with lactulose and antibiotics
is sometimes warranted (see Table 71-1).
•
Follow-up in 11 dogs for a mean period of 15 months (range of 1 week to 4.5 years)
indicated a good clinical response to dietary therapy alone. Repeated SBA concentrations
remained unchanged. How often dogs that are asymptomatic for HMD progress and develop
clinical signs is unknown.
•
Anecdotal reports suggest some severely affected dogs may develop a progressive hepatopathy
with portal hypertension and multiple extrahepatic PSSs, similar to primary hypoplasia
of the portal veins (see below).
PRIMARY PORTAL VEIN HYPOPLASIA
Primary portal vein hypoplasia (PPVH) is a congenital abnormality of portal vascular
development seen in young medium- to large-breed dogs. Small intrahepatic portal venules
are predominantly affected, although hypoplasia of the extrahepatic portal vein may
occur in 30% of dogs. Underdevelopment of the portal system leads to obstruction to
portal blood flow resulting in portal hypertension and development of multiple extrahepatic
PSSs.
•
Dogs described in the literature with idiopathic non-cirrhotic portal hypertension
have clinical and pathologic findings that resemble portal vein hypoplasia, and these
are now considered the same disorder.
•
In addition to the vascular changes, variable amounts of fibrosis may be present in
portal areas, leading to the suggestion that hepatoportal fibrosis of young dogs is
a variant of portal vein hypoplasia.
•
It has also been proposed that HMD (discussed in the previous section) is a milder
variant of PPVH and that the term HMD should be abandoned. Although histologic features
are similar, the clinical presentations of HMD and PPVH are distinctly different.
HMD is not typically associated with portal hypertension (or even clinical signs in
many cases), whereas the clinical features of PPVH are primarily related to marked
portal hypertension and subsequent portosystemic shunting. Thus, we advocate retaining
the clinical distinction between HMD and PPVH until definitive clarification is available.
Signalment and Clinical Signs
The mean age is about 2 years. Medium- or large-breed dogs appear to be at increased
risk. In one study, one unrelated and three related Doberman pinschers were reported.
Related cocker spaniels have also been described.
•
Common clinical signs include lethargy, weight loss or stunted growth, intermittent
vomiting and diarrhea, ascites, and polydipsia. Neurologic signs of hepatoencephalopathy
may occur but are inconsistent.
•
The duration of clinical signs may range from a few weeks to a year. Most dogs have
persistent signs for 2 to 3 months.
Key Point
Clinical signs of PPVH are similar to those seen in dogs with single congenital PSSs,
except that ascites is common in PPVH and rare in dogs with congenital PSS.
Diagnosis
Laboratory Evaluation
•
Laboratory findings are consistent with hepatic dysfunction and portosystemic shunting
and include microcytosis, hypoalbuminemia, decreased BUN, and mild increases in ALP
and ALT (2-4 times normal).
•
A pattern of normal to mildly increased FSBA concentrations accompanied by markedly
increased PPSBA values is consistent with portosystemic shunting.
•
Hyperammonemia and an abnormal ATT are also consistent features.
•
Ascitic fluid is typically a transudate.
Radiography and Ultrasonography
•
Survey abdominal radiographs reveal microhepatica and poor intra-abdominal contrast.
•
Microhepatica and abdominal effusion are common findings on ultrasonography. Liver
echotexture is variable. Multiple extrahepatic PSSs appear as enlarged tortuous vessels
caudal to the liver. Splenomegaly may also be detected.
•
Ultrasonography can also provide important information regarding other disorders associated
with portal hypertension, multiple PSSs, and ascites. With chronic end-stage liver
disease, the liver may be small and hyperechoic with multiple regenerative nodules.
Congenital hepatic arteriovenous fistulas appear as tortuous, anechoic tubular structures
in the liver.
Laparotomy and Contrast Portography
Multiple PSSs can be confirmed by contrast portography or at exploratory laparotomy
(or necropsy). Multiple PSSs usually appear as multiple tortuous vessels that communicate
with the caudal vena cava in the area of the left kidney.
•
The portal pressure can be measured to document portal hypertension (normal portal
pressure is 6 to 13 cm of water).
•
Patency of the portal vein should be verified (by mesenteric portography or at surgery)
since portal vein obstruction could cause similar clinical and radiographic features.
With PPVH, the extrahepatic portion of the portal vein is patent but may be underdeveloped.
•
Congenital hepatic arteriovenous fistulas can be diagnosed by celiac arteriography
or exploratory laparotomy.
Liver Biopsy
Liver biopsy is essential to confirm the diagnosis and rule out primary intrahepatic
disorders such as chronic hepatitis and cirrhosis that can cause secondary portal
hypertension and multiple acquired PSSs and that would have a different treatment
and long-term prognosis.
•
Grossly, the liver in PPVH is small and smooth or slightly irregular.
•
Microscopically, the portal veins in the portal triads are small or absent. Other
features include arteriolar hyperplasia, hepatocyte atrophy, and absence of inflammation
with variable bile duct proliferation, lymphatic distention, and portal fibrosis (which
is sometimes severe).
•
Progressive fibrosis has been documented in some dogs with PPVH undergoing serial
liver biopsies.
Treatment and Prognosis
Specific treatment for PPVH is not available. Initiate symptomatic therapy to control
the consequences of portal hypertension and portosystemic shunting, such as ascites
and HE (see Table 71-1 and “Principles of Treatment for Liver Disease”). The long-term
prognosis is variable, but some dogs can live for years on medical management. Owners
should be discouraged from electing euthanasia for affected dogs until medical management
has been tried.
•
Avoid indiscriminate dietary protein restriction since it can worsen hypoalbuminemia
and promote ascites formation. Although ascites is a common presenting sign, it may
resolve over time and diuretics can be discontinued.
•
Use H2 receptor blockers indefinitely because perforated duodenal ulcer is a potential
cause of death in PPVH (see Table 71-1).
•
Consider colchicine (0.025 mg/kg PO q24h) and ursodiol (15 mg/kg PO q24h) to prevent
progressive hepatic fibrosis.
Key Point
Surgical ligation of multiple acquired PSSs is contraindicated. Ligation may result
in fatal portal hypertension because shunts form as a protective compensatory response
to portal hypertension. Also, do not perform caudal vena caval banding.
HEPATIC ARTERIOVENOUS FISTULA
Intrahepatic arteriovenous (AV) fistulas are vascular communications between the hepatic
artery and the portal vein that result in portal hypertension, ascites, and secondary
PSSs. They occur rarely in dogs and cats.
Etiology
Intrahepatic AV fistulas may be congenital, which is most common, or acquired as a
result of abdominal trauma, hepatic surgery, hepatic neoplasia, cirrhosis, or rupture
of a hepatic artery aneurysm.
Clinical Signs
Signs are similar to those of congenital PSS and PPVH and include anorexia, lethargy,
vomiting, diarrhea, PU/PD, and encephalopathy in a young animal (usually less than
1.5 years).
Diagnosis
Historical and physical findings are similar to those in congenital PSS, with the
notable exception that marked ascites is a consistent finding with hepatic AV fistulas
but is uncommon with congenital PSS.
•
Other causes of ascites to be differentiated include hypoproteinemia and right-sided
congestive heart failure and other causes of portal hypertension and multiple PSSs,
such as PPVH (see the preceding section and Table 71-10).
•
Auscultate the abdominal wall over the area of the liver for a continuous murmur (bruit)
caused by runoff of arterial blood into the portal system.
Laboratory Evaluation
•
Laboratory abnormalities are similar to those seen in congenital PSS and PPVH and
include hypoproteinemia, normal or mildly increased serum liver enzyme activity, and
abnormal liver function tests including FSBA, PPSBA, blood ammonia, and ATT.
•
Ascitic fluid typically is a transudate.
Radiography and Ultrasonography
•
Survey radiographs show marked ascites.
•
On abdominal ultrasonography, hepatic AV fistulas appear as tortuous, anechoic tubular
structures in the liver. Multiple extrahepatic PSSs may also be identified.
Laparotomy
•
Confirm the diagnosis by celiac arteriography or exploratory laparotomy. Grossly,
AV fistulas appear as thin-walled, tortuous, pulsating vascular channels that distort
the hepatic parenchyma and elevate the overlying hepatic capsule.
Treatment
Partial hepatectomy is indicated for treatment of hepatic AV fistulas involving one
liver lobe (see Chapter 72). Dearterialization is required if multiple lobes are involved.
•
Despite resection of involved liver lobes, hepatic function may not return to normal
because of persistent shunting of portal blood through acquired PSS or concurrent
PPVH.
•
Medical management of HE with a moderately protein-restricted diet, lactulose, and
antibiotics is indicated (see Table 71-1).
HEPATOBILIARY NEOPLASIA
Neoplasms involving the liver can be categorized as primary hepatic tumors (of either
epithelial or mesodermal origin), hemolymphatic tumors, or metastatic tumors (Table
71-11
). Hemolymphatic tumors (especially lymphoma) are the most common type of neoplasia
involving the liver of cats. Metastatic tumors to the liver are most common in dogs,
especially hemangiosarcoma, islet cell carcinoma, pancreatic carcinoma, and fibrosarcoma.
Hepatic lymphoma also occurs frequently in dogs.
Table 71-11
HEPATIC NEOPLASIA IN DOGS AND CATS
Primary Hepatic Neoplasia
Epithelial Origin
Hepatocellular carcinoma
Hepatocellular adenoma
Biliary carcinoma (cholangiocarcinoma, biliary cystadenocarcinoma)
Biliary adenoma (including biliary cystadenoma)
Hepatic carcinoid
Mesodermal Origin
Hemangiosarcoma
Hemangioma
Leiomyosarcoma
Fibrosarcoma
Liposarcoma
Myxosarcoma
Osteosarcoma
Myelolipoma
Fibroma
Hemolymphatic Tumors
Lymphoma
Mast cell tumor
Myeloproliferative disorders
Plasma cell tumor
Metastatic Hepatic Neoplasia
Hemangiosarcoma
Islet cell carcinoma
Pancreatic carcinoma
Fibrosarcoma
Osteosarcoma
Transitional cell carcinoma
Intestinal carcinoma
Renal cell carcinoma
Pheochromocytoma
Thyroid carcinoma
Mammary carcinoma
Primary hepatic tumors occur infrequently in dogs and cats. They are usually of epithelial
origin and are derived from either hepatocytes or biliary epithelium. They can be
either benign or malignant.
•
A benign tumor of the hepatocytes is called a hepatocellular adenoma (or hepatoma),
and its malignant counterpart is called a hepatocellular carcinoma.
Key Point
A hepatocellular carcinoma is the most common primary hepatic tumor in dogs.
•
A benign tumor arising from the biliary epithelium is called a biliary adenoma. The
malignant form is called a biliary carcinoma. Biliary carcinomas may be intrahepatic,
extrahepatic, or within the gallbladder. The intrahepatic form is most common in dogs
and cats. Cystic forms of these tumors (cystadenocarcinoma, cystadenoma) have also
been described.
Key Point
Biliary carcinomas and adenomas are the most common primary hepatic tumors in cats.
•
Hepatocellular carcinoma and bile duct carcinoma occur in three pathologic forms:
(1) solitary, a single large mass in one liver lobe with or without smaller masses
in other lobes; (2) multifocal nodular, discrete nodules of varying sizes in several
liver lobes; and (3) diffuse, infiltration of large portions of the liver with non-encapsulated,
highly invasive neoplastic tissue. Solitary masses are most likely to be successfully
resected surgically. In dogs with hepatocellular carcinoma, approximately half of
the tumors are solitary and half are multifocal or diffuse. Biliary carcinomas are
more likely to be the multifocal or diffuse form.
Etiology
The cause of spontaneous primary hepatic neoplasms in dogs and cats is not usually
determined. Some potential causes based on reports of experimental and spontaneous
hepatic tumors include aflatoxins, nitrosamines, aramite, liver flukes (Clonorchis
spp. Platynosomum concinnum), and radioactive compounds such as strontium-90 and cesium-144.
In contrast to humans, no association with viral infections has been identified in
spontaneous tumors of dogs and cats.
Clinical Signs
Dogs and cats with hepatic neoplasia usually show vague signs of hepatic dysfunction
that often do not appear until the more advanced stages of hepatic disease.
•
The most consistent signs in dogs are anorexia, lethargy, weight loss, PU/PD, vomiting,
and abdominal distention. Other signs include jaundice, diarrhea, and excessive bleeding.
•
Signs of CNS dysfunction such as depression, dementia, or seizures can be attributed
to HE, hypoglycemia, or CNS metastases.
•
Anorexia and lethargy are the most common presenting signs in cats; ascites and vomiting
are uncommon in cats as compared with dogs.
•
When the liver is secondarily involved with metastases, the clinical signs may reflect
the primary tumor location or other metastatic sites rather than the hepatic involvement.
Diagnosis
Suspect hepatobiliary neoplasia in any older animal with clinical and biochemical
evidence of hepatic disease accompanied by hepatomegaly.
Signalment
•
Primary hepatic neoplasms are most common in dogs and cats that are older than 10
years of age.
•
Labrador retrievers were found to be disproportionately represented in one study of
dogs with biliary carcinoma.
•
Male dogs and cats may be at increased risk for hepatocellular carcinoma.
•
Male cats and female dogs have an increased risk for biliary carcinoma.
Physical Examination
•
Findings often include a cranial abdominal mass or marked hepatomegaly. Hepatomegaly
is less likely with metastatic tumors.
•
Ascites or hemoperitoneum may contribute to abdominal distention. Tumor rupture and
hemorrhage is most likely with hepatocellular adenoma, hepatocellular carcinoma, and
hepatic hemangiosarcoma.
•
Anemia and pale mucous membranes may be attributed to excessive hemorrhage from a
ruptured neoplasm or anemia of chronic disease.
•
Jaundice is a less frequent finding with hepatic tumors unless the tumor mass causes
obstruction of the common bile duct.
•
Severe weight loss and cachexia are common but nonspecific findings.
•
Myasthenia gravis causing weakness was suspected to be a paraneoplastic syndrome in
a dog with biliary carcinoma.
Laboratory Evaluation
Hematologic and biochemical findings in animals with hepatic neoplasia are not specific
and are indicative of hepatic disease and its complications.
•
Potential hematologic findings include anemia and leukocytosis. Anemia is usually
nonregenerative but may be regenerative if associated with excess bleeding or tumor
rupture. When hematopoietic or lymphoid malignancies secondarily involve the liver,
abnormal cells or pancytopenia may be detected on peripheral blood smears because
of concurrent bone marrow involvement.
•
Mild to marked increases in serum liver enzyme (ALT and ALP) activity are common in
dogs with primary hepatic tumors (60-100% of cases) but less so with metastatic neoplasia.
In contrast, most cats with non-hematopoietic hepatic neoplasms have increased serum
ALT or AST activity, but serum ALP activity is usually normal. Increased serum AST
activity may be the most sensitive indicator of metastatic hepatic disease in dogs
(80% of cases) but lacks specificity.
•
Hyperbilirubinemia appears to be a more frequent finding in dogs with metastatic neoplasia
than in those with primary hepatic tumors (59% versus 25%).
•
Hypoglycemia, sometimes severe, occasionally is noted in dogs with hepatocellular
carcinoma and less frequently with hepatocellular adenoma, leiomyosarcoma, and hemangiosarcoma.
Serum insulin concentrations are normal to decreased. Potential mechanisms of hypoglycemia
include excess utilization of glucose by the tumor, release of insulin-like factors
from the tumor, release of other substances from the tumor such as somatostatin, and
secondary hepatic parenchymal destruction with impaired glycogenolysis or gluconeogenesis.
•
Other biochemical findings are quite variable and include hypoalbuminemia, hyperglobulinemia,
and increased SBA concentrations. The magnitude of increase in SBA concentrations
in dogs with hepatic neoplasia can be quite small; SBA concentrations may be within
normal limits.
•
Although clinical evidence of impaired hemostasis is infrequent, prolongation of the
PT and APTT may be identified in dogs with hepatic neoplasia.
•
Analysis of abdominal fluid usually indicates a transudate or modified transudate;
however, neoplastic cells or a bloody effusion are occasionally noted.
•
Increased serum alpha-fetoprotein concentration may be an indicator of hepatocellular
carcinoma and biliary carcinoma in dogs. Increased alpha-fetoprotein secretion also
occurs with marked hepatic regeneration and chronic hepatitis. Alpha-fetoprotein concentrations
in cats with hepatic neoplasia have not been reported.
Radiography
•
Abdominal radiographic findings in animals with hepatic neoplasia include symmetrical
or asymmetrical hepatomegaly and ascites.
•
Perform thoracic radiographs to detect pulmonary metastases.
Ultrasonography
The diagnosis of hepatic neoplasia cannot be made based on ultrasonographic findings
alone; however, ultrasonography often reveals focal, multifocal, or diffuse changes
in hepatic echotexture.
•
Hepatocellular carcinoma usually appears as a focal hyperechoic mass.
•
Primary or secondary neoplasia and nodular hyperplasia often appear as focal or multifocal
hypoechoic or mixed echogenic lesions.
•
Target lesions, consisting of an echogenic center surrounded by a more sonolucent
rim, are often neoplastic.
•
Hepatic lymphoma is quite variable and may appear as a mild diffuse hyperechogenicity
or hypoechogenicity, multifocal hypoechoic lesions, or a mixed echogenic pattern;
alternatively, the appearance may be normal.
Liver Biopsy
Definitive diagnosis of hepatic neoplasia requires liver biopsy and histopathologic
evaluation.
•
Laparotomy is the procedure of choice for a single, large hepatic mass because excision
of the mass can also be performed concurrently.
•
Ultrasound-guided biopsy is useful to diagnose focal or diffuse hepatic involvement,
but the small size of the biopsy can make differentiation of nodular hyperplasia versus
primary hepatic neoplasia difficult. A wedge biopsy obtained at surgery is often necessary.
•
Blind percutaneous needle biopsy and FNA cytology are most useful for diagnosis of
diffuse hepatic neoplasias such as lymphoma, myeloproliferative disorder, and mast
cell tumor.
Treatment
Surgery
Surgical removal of the affected liver lobe is the treatment of choice for primary
hepatic neoplasms such as hepatocellular adenoma or carcinoma that involve a single
lobe (see Chapter 72). Early detection prior to metastasis to other liver lobes affords
the best chance for surgical control.
•
Surgical resection of a bleeding mass may provide palliative therapy despite the presence
of metastatic disease.
•
Perform a complete exploratory of the abdominal cavity for evidence of metastases,
and biopsy hepatic lymph nodes.
•
When all lobes are affected, the prognosis is poor.
Chemotherapy
Key Point
Chemotherapy is not an effective means of control for primary liver tumors in dogs
and cats.
Secondary hepatic neoplasms such as lymphoma, mast cell tumor, or myeloproliferative
disease might temporarily respond to chemotherapeutic intervention (see Chapters 26
through 28).
HEPATIC NODULAR HYPERPLASIA
Nodular hyperplasia of the liver is a common benign postmortem finding in dogs more
than 8 years of age. In one study, hepatic nodular hyperplasia was present in all
dogs older than 14 years of age. It appears to be an age-related change, but the cause
is unknown. It is not a preneoplastic disorder. The number of nodules ranges from
just a few to multiple nodules in a random distribution throughout all liver lobes.
They may be microscopic or macroscopic with distortion of the hepatic surface. With
extensive involvement, nodular hyperplasia can grossly mimic macronodular cirrhosis
or neoplasia. Occasionally, single hyperplastic nodules can become quite large and
mimic a hepatocellular adenoma both clinically and microscopically.
Clinical Signs
•
Nodular hyperplasia is not usually associated with clinical signs.
Diagnosis and Treatment
•
Hepatic nodular hyperplasia can cause mild to moderate increases in serum liver enzymes,
especially ALP activity. Other tests of liver function are typically normal.
•
These benign lesions should be considered in the differential diagnosis when hepatic
nodules are identified during ultrasonography, laparoscopy, or surgery. Ultrasonographically,
many hyperplastic nodules are not detected because they are isoechoic to adjacent
liver tissue. However, they may have a variety of echotextures and appear similar
to primary or secondary hepatic neoplasia, requiring histopathology to distinguish
these disorders. Preliminary studies suggest magnetic resonance imaging may be useful
in differentiating benign from neoplastic focal hepatic lesions in dogs.
•
FNA cytology commonly reveals vacuolated hepatocytes suggestive of fat or glycogen
accumulation. Extramedullary hematopoiesis composed of predominantly segmented and
band neutrophils may also be identified.
Key Point
Needle biopsies often do not provide adequate tissue for pathologists to diagnose
nodular hyperplasia.
•
If needle biopsy only describes vacuolated hepatocytes, the clinician may be misled
to search for causes of metabolic disease, such as hyperadrenocorticism.
•
A wedge biopsy may be necessary to differentiate nodular hyperplasia from hepatocellular
carcinoma and hepatoma.
•
No treatment is required.
HEPATIC CYSTS
Single or multiple diffuse hepatic cysts occasionally are identified in the liver
of dogs and cats, usually as incidental findings at necropsy but occasionally in the
live animal.
Etiology
Hepatic cysts can be congenital or acquired, although the distinction is often difficult
to make. In general, acquired cysts are usually solitary and congenital cysts are
often multiple.
•
Congenital polycystic disease of the liver and kidneys has been reported in Cairn
terriers and WHWTs.
•
Polycystic renal disease in cats has been associated with cystic dilation of the intrahepatic
bile ducts (see Chapter 77).
•
Acquired cysts may represent benign bile duct adenomas or biliary cystadenomas or
may occur secondary to trauma.
•
Peliosis hepatis, a vasculoproliferative disorder characterized by cystic, blood-filled
spaces in the liver, is a rare disease in dogs and cats. Infection with Bartonella
henselae was confirmed by polymerase chain reaction of liver tissue in a dog with
peliosis hepatis.
Clinical Signs
•
Most solitary hepatic cysts do not cause any clinical signs unless they compress or
displace adjacent structures. Signs are more likely to occur when congenital polycystic
disease is accompanied by dilation of the extrahepatic biliary tract.
•
Abdominal enlargement secondary to an enlarged cyst or abdominal fluid accumulation
can be a presenting sign. Peliosis hepatis may be associated with intra-abdominal
hemorrhage.
Diagnosis and Treatment
•
Hepatic cysts should be considered in the differential diagnosis of any cavitated
hepatic mass lesion detected on palpation, radiography, or ultrasonography.
•
Surgery can confirm the diagnosis and allow excision of large solitary cysts (see
Chapter 72).
FELINE INFLAMMATORY LIVER DISEASE
Inflammatory diseases of the liver are the second most common type of feline liver
disease, after hepatic lipidosis. The cause, pathogenesis, natural history, and optimal
treatment of inflammatory liver disease in cats are largely unknown. Various terms
have been used to describe these disorders, such as cholangitis or cholangiohepatitis
complex, suppurative cholangiohepatitis, non-suppurative cholangiohepatitis, lymphoplasmacytic
cholangiohepatitis, lymphocytic cholangitis, progressive lymphocytic cholangitis,
lymphocytic portal hepatitis, sclerosing cholangitis, and biliary cirrhosis.
•
Recently, the World Small Animal Veterinary Association (WSAVA) Liver Diseases and
Pathology Standardization Research Group developed a classification system with the
goal of worldwide standardization of terminology for histologic evaluation of liver
diseases in dogs and cats.
•
The term cholangitis is preferred over cholangiohepatitis because the inflammation
primarily originates in the bile ducts, and extension beyond the limiting plate into
the hepatic parenchyma (cholangiohepatitis) is not always a feature. Cholangitis is
further divided into neutrophilic (acute or chronic) or lymphocytic cholangitis. This
classification scheme will be used for the purposes of the following discussion.
Neutrophilic Cholangitis
Neutrophilic cholangitis (also called suppurative cholangitis) is the most common
form of cholangitis in cats.
•
Histologically, the lesion is characterized by neutrophilic inflammation within the
walls or lumen of bile ducts. The neutrophilic inflammation may disrupt the limiting
plate and extend into the hepatic parenchyma, causing necrosis of periportal hepatocytes.
•
Chronic neutrophilic cholangitis (previously referred to as lymphoplasmacytic or non-suppurative
cholangitis or cholangiohepatitis) is characterized by a mixed inflammatory response
(neutrophils plus lymphocytes and plasma cells). Other features of chronicity include
marked biliary hyperplasia, concentric periductal fibrosis, and bridging portal fibrosis.
Key Point
Chronic neutrophilic cholangitis may progress to biliary cirrhosis. As opposed to
the small liver in dogs with end-stage cirrhosis, cats with biliary cirrhosis typically
have hepatomegaly because of profound biliary hyperplasia.
Etiology
Key Point
Acute neutrophilic cholangitis is most likely caused by ascending bacterial infection
from the intestine into the biliary tract.
•
Bacterial organisms isolated from bile or liver tissue are primarily gram-negative
and anaerobic enteric bacteria such as E. coli (most common), Staphylococcus, alpha-hemolytic
Streptococcus, Bacillus, Actinomyces, Bacteroides, and Clostridia.
•
Chronic bacterial infections elsewhere in the body (sinusitis, splenic abscess, pyelonephritis)
and septicemia may also be associated with neutrophilic cholangitis.
•
Chronic neutrophilic cholangitis may represent a persistent bacterial infection, or
the lesion may have been initiated by bacteria but an immune-mediated response may
result in a chronic self-perpetuating disorder. Other possible causes of neutrophilic
cholangitis may include viral, toxin-induced, or drug-induced disease.
•
Infectious agents infrequently associated with lesions of cholangitis in cats include
liver flukes (see previous discussion in this chapter), Toxoplasma-like organisms,
and Hepatozoon canis.
Clinical Associations and Predispositions
Key Point
Neutrophilic cholangitis in cats is commonly associated with IBD and chronic subclinical
pancreatitis (“triaditis”).
•
In cats, the pancreatic and bile ducts join into a common channel prior to opening
into the duodenum at the sphincter of Oddi. Underlying IBD may be the key feature
that predisposes to both cholangitis and pancreatitis. Inflammation of the duodenal
mucosa could alter the normal function of the sphincter of Oddi. IBD commonly causes
vomiting in cats, and vomiting could raise the intraduodenal pressure and predispose
the patient to reflux of duodenal juice into the pancreatic and biliary systems. Since
the normal bacterial count in the proximal small intestine is higher in cats than
in dogs, it may be more likely to induce pathology in cats.
•
Congenital biliary tract malformations or biliary reconstructive surgery may also
predispose the patient to ascending bacterial infection.
•
Concurrent extrahepatic biliary tract abnormalities include cholecystitis and sludged
bile or choleliths, which may cause bile duct obstruction.
•
Cats with cholangitis may develop secondary hepatic lipidosis, possibly related to
anorexia and weight loss.
Clinical Signs
•
Acute neutrophilic cholangitis causes acute onset of vomiting, anorexia, lethargy,
and weight loss.
•
Chronic neutrophilic cholangitis causes intermittent or persistent vomiting, anorexia,
lethargy, and weight loss lasting weeks to years. Signs of HE, ascites, and excessive
bleeding are uncommon unless chronic neutrophilic cholangitis has progressed to biliary
cirrhosis.
Diagnosis
Suspect neutrophilic cholangitis in any cat with non-hemolytic jaundice, especially
if accompanied by fever (although not all cases are febrile). Definitive diagnosis
requires liver biopsy to distinguish this disease from other hepatic disorders, such
as hepatic lipidosis, hepatic FIP, and neoplasia.
History and Physical Examination
•
Cats with acute neutrophilic cholangitis typically have an acute clinical presentation
of short duration (<5 days). Young cats appear to be at increased risk. A history
of antibiotic-responsive illness is common.
•
In contrast, cats with chronic neutrophilic cholangitis are usually middle age or
older.
•
Physical examination findings include fever, jaundice, abdominal pain, hepatomegaly,
and dehydration. Fever and abdominal pain are more likely with acute cholangitis.
Key Point
Ascites may be present if chronic neutrophilic cholangitis has progressed to cirrhosis.
Laboratory Evaluation
•
For both acute and chronic disease, liver enzyme elevations are variable. Although
cholangitis is typically considered a cholestatic disorder, the ALP may be normal,
especially in the acute form. This may occur because ALP is an induced enzyme with
a lag period before it increases in the serum.
•
Increased ALT is a more consistent finding and probably indicates inflammation is
extending beyond the portal area and limiting plate, resulting in hepatocellular necrosis
(cholangiohepatitis).
•
Hyperbilirubinemia and bilirubinuria are common in both the acute and the chronic
forms of cholangitis. FSBAs and PPSBAs are consistently increased.
Key Point
Neutrophilia with a left shift is most frequent in cats with the acute form of neutrophilic
cholangitis, whereas hyperglobulinemia is a frequent finding in cats with the chronic
form.
•
Cats with chronic neutrophilic cholangitis frequently have a mild nonregenerative
anemia. A coagulopathy may occur secondary to vitamin K malabsorption, hepatocyte
failure, or DIC. Hypoalbuminemia, decreased BUN, and hyperammonemia suggest advanced
disease.
•
Consider evaluating serum cobalamin level (B12) and feline pancreatic lipase immunoreactivity
because of the association of neutrophilic cholangitis with IBD and pancreatitis (see
Chapters 69 and 73).
•
In endemic tropical locations, rule out liver flukes by fecal exams or a therapeutic
trial of praziquantel.
Radiography and Ultrasonography
•
Abdominal radiographs may reveal a liver that is normal or increased in size. Radiopaque
choleliths may also be detected.
•
Ultrasonographically, cats with neutrophilic cholangitis have no detectable parenchymal
abnormalities, a diffuse hepatic hypoechogenicity, or a coarse or nodular liver texture.
This helps differentiate cholangitis from lipidosis, which causes hyperechogenicity
of the liver.
•
Ultrasonography is also useful to evaluate for concurrent abnormalities of the biliary
tract and pancreas, such as common bile duct obstruction, cholelithiasis, sludging
of bile, cholecystitis, and pancreatitis.
Liver Biopsy
Definitive diagnosis requires liver biopsy to distinguish neutrophilic cholangitis
from other hepatic disorders, such as hepatic lipidosis, hepatic FIP, and neoplasia.
•
If concurrent biliary obstruction occurs, obtain a liver biopsy specimen and aerobic
and anaerobic bile cultures at laparotomy during surgical relief of the obstruction
(see Chapter 72). Duodenal and pancreatic biopsies are also indicated. At laparotomy,
the gallbladder and common bile duct frequently are thickened, firm, and distended.
Inspissated bile and choleliths may be present.
•
In the absence of obstruction, ultrasound-guided liver biopsy is adequate for diagnosis
in most cats. Laparoscopic liver biopsy provides larger tissue samples than those
obtained with ultrasound guidance. Perform an aerobic and anaerobic bacterial culture
of liver tissue (or bile obtained by cholecystocentesis).
•
Cytologic evaluation of impression smears of bile or liver tissue has been recommended
since bacteria are more easily detected cytologically than histopathologically.
Treatment
General Supportive Care
•
Treat neutrophilic cholangitis with fluid therapy, nutritional support, and supplementation
of potassium and B vitamins (see Table 71-1).
•
Give vitamin K1 (0.5-1.5 mg/kg SC or IM q12h for three doses) if hemostasis evaluation
suggests vitamin K deficiency.
•
Avoid dietary protein restriction unless overt signs of HE are present. If concurrent
IBD is suspected, dietary modifications may include commercial GI or novel protein
diets (see Chapter 69).
Antibiotics
Key Point
Antibiotics are the primary therapy for acute neutrophilic cholangitis. They are also
used in the initial therapy of chronic neutrophilic cholangitis prior to initiating
glucocorticoid therapy, in order to eliminate any bacterial component.
If possible, base the choice of antibiotic on culture and sensitivity testing results;
otherwise, consider the following recommendations: Continue antibiotics for a minimum
of 6 to 8 weeks. In some cats with persistent elevation of serum bilirubin and liver
enzymes, continue antibiotic therapy for 3 to 6 months.
•
For initial treatment, consider ampicillin (20–40 mg/kg PO, IV, IM or SC q8h), amoxicillin
(10–20 mg/kg PO, IV, or SC q8-12h), amoxicillin-clavulanate (62.5 mg/cat PO q12h),
or a cephalosporin. Combine one of these with metronidazole (7-10 mg/kg PO q12h) for
broader spectrum against anaerobes, as well as modulation of cell-mediated immunity
and treatment of concurrent IBD.
•
For refractory cases, use enrofloxacin (2.5 mg/kg PO, IM, or IV q12h) or a 1-week
course of an aminoglycoside.
Prednisolone
•
Prednisolone is recommended for treatment of cats with chronic neutrophilic cholangitis
because of its anti-inflammatory and immunosuppressive properties. However, efficacy
of corticosteroids for treatment of chronic neutrophilic cholangitis has never been
proven.
•
Give prednisolone, 2 to 4 mg/kg q24h (once daily or divided), until clinical remission,
then taper over 6 to 8 weeks to 1 to 2 mg/kg once every other day.
Hepatoprotective Therapy
•
Give ursodiol (Actigall, Ciba Geneva) to all cats with neutrophilic cholangitis once
extrahepatic biliary obstruction is eliminated because of its hepatoprotective (anti-inflammatory,
immunomodulatory, and antifibrotic effects) properties (see Table 71-2). It appears
to be well tolerated and safe in cats.
•
Consider other hepatoprotectants, such SAMe or vitamin E (see Table 71-2).
Surgery
Surgical intervention is indicated if biliary obstruction (from sludged bile or choleliths)
is detected on ultrasound examination. Surgical procedures for the biliary tract are
described in Chapter 72. Use supportive care (including treatment of coagulopathy)
for stabilization prior to the procedure. Indications for surgical intervention in
the treatment of neutrophilic cholangitis include the following:
•
Biliary decompression for extrahepatic biliary obstruction
•
Sludge removal and bile duct and gallbladder irrigation
•
Cholelith removal
•
Cholecystectomy for necrotizing cholecystitis
Biliary diversion techniques such as cholecystoduodenostomy and cholecystojejunostomy
are most commonly performed when biliary obstruction is present; however, the mortality
rate ranges from 23% to 32% within 6 months of surgery. Cats with biliary surgery
that did survive had repeated intermittent bouts of fever, vomiting, and anorexia,
which responded to antibiotics. Consequently, if biliary obstruction is present, procedures
to establish patency of the biliary system without performing a biliary diversion
surgery appear preferable.
Prognosis
Cats with acute neurophilic cholangitis may have a complete recovery after treatment
with antibiotics. However, some of these cats progress to the chronic form.
Key Point
Many cats with chronic neutrophilic cholangitis continue to have smoldering disease
requiring continued treatment for months or years.
Cats with neutrophilic cholangitis that survive less than 1 year commonly have concurrent
diseases that probably contribute to their death.
Lymphocytic Cholangitis
Lymphocytic cholangitis (also called progressive lymphocytic cholangitis or cholangiohepatitis)
is believed to be a distinct form of cholangitis in the cat. It appears to be rare
in the United States but has been well described in Europe. An immune-mediated pathogenesis
has been suggested. The histologic lesion is characterized by moderate to marked infiltration
of small lymphocytes in portal areas, with variable portal fibrosis and bile duct
proliferation. As the lesion progresses, the number of lymphocytes decreases but marked
fibrosis disrupts the hepatic architecture.
•
Persian cats may have a genetic predisposition.
•
The most common clinical features are ascites, icterus, generalized lymphadenopathy,
and hyperglobulinemia in young cats.
•
Concurrent abnormalities of the extrahepatic biliary system, intestine, or pancreas
are not seen.
•
Treatment options for lymphocytic cholangitis are similar to those described previously
for chronic neutrophilic cholangitis.
Lymphocytic Portal Hepatitis
Lymphocytic infiltration of portal areas without bile duct involvement or periportal
necrosis is termed lymphocytic portal hepatitis. It is believed to be a nonspecific
reaction and not a primary disease process.
•
Lymphocytic portal hepatitis is a common finding in liver biopsies of cats older than
10 years of age (up to 82% of cats). In contrast, only 10% of cats younger than 10
years had this finding. It is not associated with IBD or pancreatitis. Liver enzyme
elevations may or may not be present.
•
Whether or not treatment with corticosteroids is indicated is unclear. The mean survival
time in 23 cats with lymphocytic portal hepatitis was 29 months; 72% of these cats
were not treated with corticosteroids.
•
In cats with marked lymphocytic infiltration in the portal areas, it can be difficult
to distinguish lymphocytic portal hepatitis or lymphocytic cholangitis from well-differentiated
hepatic lymphoma.
CHOLECYSTITIS
Cholecystitis, or inflammation of the gallbladder, is a clinical problem that occurs
uncommonly in both dogs and cats. Cholecystitis may be associated with cholangitis,
cholangiohepatitis, gallbladder mucocele, cholelithiasis, and choledocholithiasis.
Acute necrotic cholecystitis in dogs frequently is complicated by rupture of the gallbladder
and septic bile peritonitis.
Etiology
•
Bacteria appear to play an important role in cholecystitis. An enteric origin of bacteria
seems most likely, because isolates are usually aerobic gram-negative bacteria (especially
E. coli, but also Klebsiella, Pseudomonas, and Salmonella spp.) or anaerobes (Clostridium
spp.). Intestinal bacteria may be refluxed into the gallbladder, or they may be blood-borne
from the hepatic circulation.
•
Campylobacter jejuni bacteremia and acute cholecystitis have been documented in dogs.
Although the GI tract was suspected to be the source of the bacteria, diarrhea was
not a presenting clinical sign.
•
Gas-producing organisms, such as E. coli and Clostridium, can cause emphysema of the
gallbladder wall. Emphysematous cholecystitis is recognized most frequently in diabetic
dogs.
•
Cholelithiasis can predispose the patient to cholecystitis by obstructing the cystic
duct, causing gallbladder overdistension and stasis, which enables proliferation of
anaerobic organisms.
•
Anatomic malformations of the gallbladder, biliary obstruction from any cause, and
biliary surgery also predispose the patient to biliary infections.
Clinical Signs
•
Signs include anorexia, lethargy, fever, abdominal pain, hepatomegaly, vomiting, diarrhea,
and jaundice.
•
Acute rupture of the gallbladder with septic bile peritonitis causes abdominal distension,
collapse, and septic shock (see also Chapter 76).
•
Signs may be acute or chronic and persistent or episodic.
Diagnosis
Differentiate cholecystitis from other cholestatic hepatobiliary disorders that have
fever, inflammation, and similar clinical findings, such as acute pancreatitis or
pancreatic abscess, cholangiohepatitis, cholelithiasis, hepatic abscess, and septicemia
or endotoxemia.
Physical Examination
Physical examination findings include fever, cranial abdominal pain, jaundice, and
shock (bile or septic peritonitis).
Laboratory Evaluation
•
Laboratory findings are characteristic of severe cholestatic hepatobiliary disease,
including hyperbilirubinemia, markedly increased ALP and GGT activity, increased ALT
activity, increased SBA concentrations, and hypercholesterolemia.
•
Other findings suggestive of inflammation or sepsis are neutrophilia with a left shift
and hypoglycemia.
•
Increased amylase and lipase levels have been reported in dogs with cholecystitis
in the absence of clinical pancreatitis.
•
Prolonged PT and APTT occur if chronic biliary obstruction causes vitamin K malabsorption.
•
Other laboratory findings reflect dehydration and electrolyte and acid-base imbalances
secondary to vomiting, dehydration, and sepsis.
•
If cholecystitis is complicated by rupture of the gallbladder or biliary tract, abdominal
fluid analysis is consistent with septic bile peritonitis. For a general discussion
of peritonitis, see Chapter 76.
Radiography and Ultrasonography
•
Potential radiographic findings include cholecystolithiasis, emphysema of the gallbladder
wall, and, if perforation occurs, abdominal effusion.
•
Ultrasonographic findings include distension of the gallbladder and cystic duct, increased
echogenicity or hypoechoic thickening of the gallbladder wall, cholecystoliths, and
increased echogenicity of gallbladder bile with or without bile sludge. Small amounts
of abdominal effusion not evident on survey radiographs may also be identified.
•
Ultrasonographic-guided percutaneous cholecystocentesis has been recommended to obtain
bile for cytology and culture (aerobic and anaerobic). Perform blood cultures to isolate
bacteria associated with bacteremia and acute cholecystitis. Culture for C. jejuni
requires selective culture techniques.
Surgery
•
Findings at exploratory surgery include thickening, necrosis, and rupture of the gallbladder,
localized or generalized peritonitis, and calculi or inspissated bile in the gallbladder,
cystic duct, or bile duct. Previous gallbladder rupture may be associated with omental
or hepatic adhesions.
•
Obtain aerobic and anaerobic cultures of the gallbladder mucosa and bile.
•
Histologic examination of the gallbladder reveals vary-ing degrees of necrosis, inflammation,
and fibrosis.
Treatment
•
Give parenteral vitamin K1 prior to surgery to correct coagulopathy (see Table 71-1).
•
Administer antibiotic therapy effective against aerobic gram-negative and anaerobic
bacteria, as described previously for bacterial cholangitis (see under “Feline Inflammatory
Liver Disease”). For initial treatment of acute cholecystitis with bacteremia or necrotic
cholecystitis with septic peritonitis, combine amoxicillin or cephalosporin with an
injectable aminoglycoside or enrofloxacin. Long-term treatment (6-8 weeks) with an
oral antibiotic is indicated.
•
Cholecystectomy is required in most cases (see Chapter 72). Surgically manage complications
such as biliary obstruction, cholelithiasis, inspissated bile, and abdominal drainage
for septic bile peritonitis (see Chapter 76).
Postoperative Care and Complications
•
Correct all fluid, electrolyte, and acid-base imbalances.
•
Common postoperative complications include vomiting, diarrhea, anorexia, hypoproteinemia,
and hypokalemia.
Prognosis
•
Death usually is attributed to sepsis and peritonitis.
•
If the animal survives the immediate postoperative period, the long-term prognosis
is good and the recurrence of biliary or hepatic disease is unlikely.
GALLBLADDER MUCOCELE
Gallbladder mucocele is an abnormal accumulation of mucus in the gallbladder lumen
accompanied by cystic mucosal hyperplasia of the gallbladder mucosa.
•
Dogs with gallbladder mucoceles can be asymptomatic early in the course of disease.
Clinical and biochemical abnormalities occur when mucoceles are complicated by secondary
bacterial infection, extrahepatic biliary obstruction, or marked distention of the
gallbladder leading to ischemic necrosis, gallbladder rupture, and bile peritonitis.
Key Point
Gallbladder rupture is a common life-threatening complication of mucocele.
•
The incidence of gallbladder mucocele appears to be increasing, and it is one of the
most common causes of extrahepatic biliary tract disease in dogs.
Etiology
The cause of gallbladder mucocele formation in dogs is unknown.
•
In humans, gallbladder mucoceles form secondary to functional or mechanical biliary
obstruction associated with cholecystitis, cholangitis, or cholelithiasis.
•
In dogs, it is not clear whether gallbladder mucoceles are a primary gallbladder disorder
or whether biliary obstruction leads to increased mucus secretion. However, predisposing
disorders causing mechanical biliary obstruction (infiltrative disease of cystic duct,
cholelithiasis) are not typically identified. A primary motility disorder of the gallbladder
with delayed gallbladder emptying could potentially predispose the patient to functional
gallbladder obstruction.
•
A primary bacterial or inflammatory disorder of the gallbladder and biliary tract
appear unlikely, since aerobic and anaerobic cultures are frequently negative and
gallbladder inflammation is inconsistent and, if present, occurs in association with
gallbladder wall necrosis.
•
Dogs with hyperadrenocorticism (or receiving corticosteroids) appear to be at increased
risk for mucocele formation. Experimentally, progestational compounds can induce lesions
of gallbladder cystic mucosal hyperplasia in dogs.
•
Acute pancreatitis may be a concurrent finding or a postoperative complication of
gallbladder mucocele surgery.
•
Increased recognition of gallbladder mucoceles in dogs may suggest a role for nutritional
or environmental factors, or it may be the result of increased use of abdominal ultrasonography.
Clinical Signs
•
Common clinical signs include anorexia, lethargy, vomiting, icterus, diarrhea, weight
loss, PU/PD, abdominal discomfort, and abdominal distention.
•
Some dogs with gallbladder mucocele are clinically (and biochemically) normal.
Diagnosis
History and Physical Exam
•
Gallbladder mucocele appears to be more likely in older (>10 years) small to medium-sized
dogs. No sex predilection is apparent. Cocker spaniels and Shetland sheepdogs appear
to be at increased risk.
•
Signs are usually acute to subacute and less than 3 weeks in duration.
•
Physical examination findings include depression, weakness, lethargy, abdominal pain,
icterus, fever, hepatomegaly, tachypnea, and tachycardia. Most dogs with gallbladder
rupture have abdominal pain.
•
Physical examination may be unremarkable.
Laboratory Evaluation
•
Hematologic findings often include leukocytosis (mature neutrophilia or neutrophilic
with left shift) and monocytosis.
•
Increased liver enzyme activity (ALP, ALT, AST, and GGT) and hyperbilirubinemia are
common biochemical features. Other less consistent findings include hypercholesterolemia,
hyperglobulinemia, hypoalbuminemia, and azotemia.
•
Values for serum ALP and ALT, total bilirubin, and WBC count are higher in dogs with
gallbladder mucocele and secondary rupture than in dogs without rupture. In one study,
all dogs with high venous lactate concentrations had a ruptured gallbladder.
Key Point
Biochemical findings may be normal in some dogs with early gallbladder mucocele formation
detected ultrasonographically.
Ultrasonography
Key Point
The ultrasonographic appearance of a gallbladder mucocele is characteristic. The gallbladder
bile is echogenic and organized in a stellate or finely striated, “kiwi fruit” pattern.
•
As opposed to biliary sludge, a gallbladder mucocele is not gravity dependent.
•
Ultrasonographic findings suggestive of secondary gallbladder rupture include loss
of gallbladder wall continuity, hyperechoic fat or fluid around the gallbladder, free
abdominal fluid, and striated or stellate echogenic material outside the gallbladder.
•
Additional findings may include extrahepatic biliary obstruction and pancreatitis.
Laparotomy
•
At surgery, the gallbladder is often markedly distended and firm, with dark serosal
discoloration.
•
The gallbladder contents appear as a shiny, greenish-black to brown gelatinous material
that often has a striated pattern.
•
If gallbladder rupture has occurred, abdominal contamination with mucocele contents
is present.
•
Perform a liver biopsy and aerobic and anaerobic bacterial cultures of bile. Perform
cholecystectomy, and flush the bile ducts to remove any residual gelatinous material
(see Chapter 72). Submit the excised gallbladder for histopathologic examination.
Histopathology
•
Histopathology of the gallbladder reveals cystic mucosal hyperplasia. Cholecystitis
is an inconsistent finding and is most likely associated with areas of ischemic necrosis
and rupture.
•
Liver biopsy findings are nonspecific and include mild to moderate portal hepatitis
and fibrosis with bile duct proliferation and vacuolar hepatopathy.
Treatment
Key Point
Cholecystectomy is the treatment of choice of gall bladder mucocele.
•
Perform cholecystectomy when a gallbladder mucocele is diagnosed ultrasonographically
in a dog with clinical and biochemical evidence of hepatobiliary disease. If gallbladder
rupture is suspected, emergency surgery is warranted (see “Biliary Rupture”).
•
Also perform cholecystectomy for treatment of gallbladder mucoceles in dogs without
clinical and biochemical abnormalities, since gallbladder rupture is a life-threatening
and unpredictable complication.
•
Preoperatively stabilize the patient with fluid therapy and antibiotics. Give broad-spectrum
antibiotic therapy (amoxicillin combined with enrofloxacin) to treat secondary bacterial
infections of the biliary tract or if gallbladder rupture is suspected. Base antibiotic
therapy on results of culture and sensitivity testing of bile or peritoneal fluid
(rupture) when possible.
•
If a coagulopathy is detected, give vitamin K1 for 24 to 48 hours prior to surgery
(see Table 71-1).
•
When gallbladder mucocele is diagnosed in an asymptomatic dog and concurrent systemic
disorders or risk of anesthesia precludes surgery, consider antibiotic therapy to
control secondary bacterial infections and monitor biochemically and ultrasonographically.
•
The role of choleretics such as ursodiol to prevent progression of mucocele formation
is unclear. Although ursodiol can increase the watery component of bile, it is contraindicated
if biliary obstruction is present.
Prognosis
The long-term prognosis after cholecystectomy appears to be excellent if the dog survives
the postoperative period.
•
Postoperative complications include sepsis and biliary infection, leakage at the surgery
site with bile peritonitis, obstruction of the common bile duct by residual mucus,
and acute pancreatitis.
•
Mortality rates are similar in dogs with gallbladder rupture and prompt surgical intervention
compared with dogs without gallbladder rupture.
•
Although a dramatic decrease in liver enzymes and bilirubin occurs after surgery,
some dogs have mild, persistent liver enzyme elevations that may be due to a concurrent
chronic inflammatory hepatopathy.
CHOLELITHIASIS
Cholelithiasis occurs infrequently in dogs and cats. Choleliths may be present in
the gallbladder (cholecystolithiasis), common bile duct (choledocholithiasis), or
rarely, hepatic and lobar ducts. Most choleliths in dogs and cats consist of insoluble
bile pigments. Minor components such as calcium, bile salts, protein, magnesium, phosphorus,
iron, carbonate, and cholesterol also have been identified. Cholesterol choleliths,
the most common type of stone in humans, are less likely to form in dogs because the
cholesterol content of dog bile is lower than that in humans, and dogs have a better
capacity for maintaining biliary cholesterol in solution. Little is known about the
cholesterol content of cat bile, but cholesterol choleliths have been reported.
Etiology
The cause of spontaneous cholelithiasis in dogs and cats often cannot be determined.
It is generally believed that gallstone formation requires initial nidus formation,
retention of particles in the gallbladder, and then sustained growth of the cholelith.
The following factors may be important in the development of pigment stones:
•
Bile stasis and sludged bile are primarily composed of mucin, which may act as a nidus
for cholelith formation by subsequently binding calcium bilirubin pigments and cholesterol
crystals.
•
Cholecystitis and cholangitis can be associated with cholelithiasis, especially in
cats (see “Feline Inflammatory Liver Disease”). It is difficult to determine whether
choleliths were formed as a consequence of bile stasis, inflammation, and bacterial
infection or whether cholelithiasis initiated the inflammation, which led to secondary
biliary stasis and infection.
•
Bacteria such as E. coli contain beta-glucuronidase, which can deconjugate bilirubin
to a less soluble form that precipitates with calcium.
•
Dietary factors are unlikely with balanced diets; however, dogs fed an experimental
diet that is low in protein and fat, high in carbohydrates, and supplemented with
cholesterol will form pigment stones. This diet is deficient in taurine, which may
contribute to cholelithiasis by precipitating bile acids.
Clinical Signs
Dogs and cats with cholelithiasis often are asymptomatic. Clinical signs are most
likely when cholelithiasis is complicated by bacterial infection, extrahepatic bile
duct obstruction, perforation of the gallbladder or bile ducts, or secondary hepatic
involvement (cholangiohepatitis or biliary cirrhosis).
•
Common signs include vomiting, anorexia, weakness, PU/PD, jaundice, weight loss, and
dehydration.
•
Signs may be acute or chronic and intermittent or persistent. An acute onset is most
likely with sudden obstruction of the cystic or common bile duct by the cholelith
or rupture of the gallbladder.
Diagnosis
Although it is uncommon, consider cholelithiasis in the differential diagnosis of
any dog or cat with cholestatic hepatobiliary disease.
History and Physical Examination
•
Aged, small-breed female dogs appear to be at increased risk.
•
A long-standing history (months to years) of intermittent jaundice and vomiting is
present in some affected animals.
•
Physical examination may be unremarkable, or findings may include jaundice, abdominal
discomfort, hepatomegaly, fever, and abdominal distention. Fever is usually indicative
of concurrent biliary bacterial infection or septic or bile peritonitis. Abdominal
distension due to fluid accumulation is seen with secondary rupture of the biliary
tract.
•
Excessive bleeding may be noted with chronic common bile duct obstruction.
•
Acholic feces are indicative of complete bile duct obstruction.
Laboratory Evaluation
Laboratory findings may be unremarkable. Biochemical evaluation of symptomatic patients
is not specific for cholelithiasis but is indicative of cholestatic hepatobiliary
disease.
•
Findings include moderate to marked increases in serum ALP and GGT activity and in
cholesterol, SBA, and total serum bilirubin concentrations. Serum ALT activity usually
is increased, indicating secondary hepatocyte damage associated with severe cholestasis
or cholangiohepatitis.
•
Potential hematologic findings include neutrophilia with a left shift, usually indicating
bacterial cholangiohepatitis or cholecystitis or complications such as a ruptured
gallbladder. A mild, nonregenerative anemia is common.
•
With chronic extrahepatic bile duct obstruction, coagulation tests may be affected
by vitamin K malabsorption.
•
With biliary rupture, abdominocentesis reveals bile peritonitis.
Radiography
•
On routine abdominal radiographs, choleliths may appear as radiopaque densities in
the area of the gallbladder or bile ducts. However, pigment stones are usually radiolucent
unless they contain calcium. Hepatomegaly is common.
•
Other findings are determined by the presence of complications such as obstruction
(a distended gallbladder), emphysematous cholecystitis (gas density in the area of
the gallbladder), and peritonitis (loss of abdominal detail).
Ultrasonography
•
Ultrasonography detects both radiolucent and radiopaque choleliths as hyperechoic
densities in the gallbladder and bile ducts. Choleliths are differentiated from mural
masses by the presence of acoustic shadowing and movement of the density with changes
in position of the animal.
•
Inspissated or sludged bile also appears in the gallbladder as an echogenic substance,
but sludge does not cause acoustic shadowing. Sludged bile may indicate biliary stasis
but can also be seen in sick, anorexic animals without clinical biliary tract disease.
•
Complications of cholelithiasis can be identified ultrasonographically, such as distention
of the gallbladder and bile ducts with cystic or common bile duct obstruction, thickening
of the biliary tract associated with inflammation, abdominal fluid accumulation with
rupture of the gallbladder, and absence of the gallbladder.
Key Point
Because the majority of choleliths do not cause clinical signs, surgical removal may
not always be warranted.
Laparotomy
Perform exploratory laparotomy for definitive diagnosis and treatment of cholelithiasis
(see Chapter 72). Pigment choleliths usually are greenish-brown to black and may be
single or multiple. Perform the follow-ing diagnostic and therapeutic procedures during
exploratory laparotomy:
•
Evaluate the patency of the gallbladder and bile ducts.
•
Remove choleliths for chemical analysis and bacterial culture.
•
Identify and repair secondary biliary rupture.
•
Collect samples of affected tissue (liver, gallbladder) and bile for aerobic and anaerobic
bacterial culture and biopsy.
Histopathologic Evaluation
Histopathologic changes in the gallbladder, bile ducts, and liver may be absent with
uncomplicated cholelithiasis. However, mild cholangitis (cholangiohepatitis) and cholecystitis
are common.
Treatment and Prognosis
•
Institute supportive therapy to correct fluid, electrolyte, and acid-base imbalances
prior to surgery. Feed a well-balanced diet.
•
If a coagulopathy is detected, give vitamin K1 for 24 to 48 hours prior to surgery
(see Table 71-1).
•
Administer systemic antibiotics in animals with inflammatory biliary tract disease
and cholelithiasis. Ideally, base the choice of antibiotic on culture and sensitivity
testing of bile and hepatic tissue obtained at surgery. See the discussion of antibiotic
therapy of biliary infections under “Neutrophilic Cholangitis.”
•
Management of complications of cholelithiasis, such as biliary obstruction or rupture,
is discussed in the next sections. Surgery of the biliary tract is discussed in Chapter
72.
•
Little is known about the likelihood of recurrence of cholelithiasis in dogs and cats,
but if the underlying mechanism of cholelith formation is not reversed, recurrence
is possible.
EXTRAHEPATIC BILIARY OBSTRUCTION
Extrahepatic biliary obstruction of the common bile duct or large hepatic ducts interrupts
bile flow into the intestine.
Etiology
Biliary obstruction can be a complication of primary biliary tract disorders such
as gallbladder mucocele, cholelithiasis, or biliary tumors or can be caused by extrahepatic
disorders such as pancreatic fibrosis and pancreatic or duodenal masses (Table 71-12
).
Table 71-12
CAUSES OF EXTRAHEPATIC BILIARY OBSTRUCTION
Cholelithiasis
Inspissated (sludged) bile
Gallbladder mucocele
Cholangitis and cholecystitis
Acute pancreatitis, pancreatic abscess, pancreatic fibrosis
Biliary, hepatic, pancreatic or duodenal neoplasia
Biliary stricture
Biliary hematoma
Liver flukes
Diaphragmatic hernia with entrapment of the gallbladder
Clinical Signs
•
Signs of biliary obstruction include anorexia, vomiting, jaundice, weight loss, abdominal
pain, diarrhea, acholic feces, and excessive bleeding.
•
Diarrhea and steatorrhea are characterized by tan-colored feces and are attributed
to failure to secrete bile acids, which results in malabsorption of fat and fat-soluble
vitamins such as vitamin K.
•
With prolonged extrahepatic biliary obstruction, vitamin K malabsorption and the subsequent
decreased synthesis of vitamin K–dependent factors results in a coagulopathy.
•
With complete biliary obstruction, the feces may become clay colored (acholic) because
of a lack of bile pigments.
Diagnosis
The diagnostic strategy is to identify that biliary obstruction is present and then
to identify the underlying cause of obstruction.
Physical Examination
•
Findings include jaundice and hepatomegaly due to bile engorgement of the liver.
•
A firm, distended gallbladder occasionally is palpated.
•
Other findings are dependent on the underlying cause of obstruction, such as palpation
of an abdominal mass (pancreatic or biliary neoplasia) and abdominal pain (acute pancreatitis,
peritonitis).
•
Fever may suggest bacterial cholecystitis or cholangiohepatitis, biliary rupture with
peritonitis, pancreatitis, or pancreatic abscess.
Laboratory Evaluation
•
Biochemical findings reflect marked cholestasis, including increased serum concentrations
of ALP, GGT, cholesterol, bile acids, and bilirubin. Unfortunately, biochemical findings
cannot distinguish whether cholestasis is caused by intrahepatic or extrahepatic mechanisms.
In general, values for total bilirubin and ALP activity tend to be higher with extrahepatic
biliary obstruction. Serum ALT and AST activity are concurrently increased due to
secondary hepatic damage.
•
On the CBC, a mild neutrophilia and mild, non-regenerative anemia are common. Neutrophilia
with a left shift suggests the possibility of acute pancreatitis or abscess, bacterial
cholangitis or cholecystitis, or biliary rupture.
•
Findings on urinalysis include bilirubinuria and absence of urobilinogen.
•
With vitamin K malabsorption, findings include prolonged PT, APTT, and activated clotting
time. Platelet function defects have also been documented in dogs with biliary obstruction.
Radiography
Abdominal radiography is frequently non-diagnostic.
•
Occasionally, a large, fluid-filled gallbladder can be seen superimposed over the
liver. The liver may be normal to increased in size. Chronic biliary obstruction in
dogs may lead to biliary cirrhosis and microhepatica.
•
Other radiographic findings depend on the underlying cause of obstruction and may
include cholelithiasis, emphysematous cholecystitis, pancreatitis, and mass lesions.
Ultrasonography
Ultrasonography is helpful to confirm extrahepatic biliary obstruction and to evaluate
the underlying cause.
•
In normal dogs, the cystic duct, common bile duct, and intrahepatic ducts are not
visible. The common bile duct may be visible in some normal cats but is usually ≤
4 mm. A common bile duct > 5 mm is suggestive of extrahepatic biliary obstruction.
•
With biliary obstruction, the biliary system, including the gallbladder, cystic duct,
common bile duct, and intrahepatic ducts, becomes progressively dilated. The earliest
detectable change is distension of the gallbladder and cystic duct, which occurs within
24 hours. By 48 hours, the common bile duct also is distended. Distention of intrahepatic
ducts is not detected until 4 to 7 days after obstruction. Dilated hepatic biliary
ducts are differentiated from hepatic and portal veins by their tortuosity and irregular
branching patterns.
•
Ultrasonography may identify underlying causes of biliary obstruction such as gallbladder
mucocele, cholelithiasis, pancreatitis, or mass lesions.
Hepatobiliary Scintigraphy
Hepatobiliary scintigraphy may be used to confirm biliary obstruction but is available
only at tertiary referral centers.
Laparotomy
Exploratory laparotomy usually is required to confirm extrahepatic biliary obstruction
and to identify the underlying cause. Perform the following diagnostic procedures:
•
Evaluate bile duct and gallbladder patency.
•
Identify location and cause of obstruction.
•
Evaluate for evidence of secondary rupture of the biliary tract.
•
Collect a sample of bile for aerobic and anaerobic bacterial cultures.
•
Perform a liver biopsy.
Treatment
Surgery
Specific therapy requires surgery to correct the underlying cause of obstruction (see
Chapter 72).
•
Prior to surgery, stabilize the patient with fluid therapy. Give vitamin K1 parenterally
for 24 to 48 hours prior to surgery to correct a coagulopathy.
•
With complete biliary obstruction, antibiotics do not enter the bile.
Medical Therapy
If biliary obstruction occurs secondary to acute pancreatitis, manage the pancreatitis
medically (see Chapter 73) and reserve surgery for those patients in which biliary
obstruction does not resolve with resolution of pancreatic inflammation.
BILIARY RUPTURE
Leakage of bile into the abdominal cavity results in chemical peritonitis that can
be complicated by sepsis.
Etiology
•
Biliary tract rupture is commonly caused by blunt or sharp abdominal trauma from automobile-induced
injuries, gunshot injuries, and bite wounds. Rupture of the common bile duct is most
likely with blunt abdominal trauma.
•
Gallbladder mucocele and necrotizing cholecystitis can be associated with gallbladder
rupture.
•
Other causes of gallbladder rupture include cholelithiasis, biliary neoplasms, gallbladder
infarction, and iatrogenic puncture during percutaneous liver biopsy.
Clinical Signs
•
When gallbladder rupture occurs secondary to gallbladder mucocele, cholecystitis or
cholelithiasis, acute onset of anorexia, vomiting, diarrhea, jaundice, abdominal pain,
fever, and shock may occur.
•
Signs of biliary duct rupture secondary to trauma tend to be chronic and develop more
slowly than with rupture of the gallbladder. With traumatic biliary rupture, early
signs such as abdominal pain and vomiting are frequently overshadowed by more immediate
signs of shock, fractures, and other injuries.
•
Other signs, such as anorexia, listlessness, weight loss, jaundice, ascites, and acholic
feces, do not occur until days or weeks following the traumatic event.
Diagnosis
History and Physical Examination
•
A history of recent abdominal trauma and progressive jaundice and abdominal distention
suggests the possibility of biliary rupture.
•
Physical examination findings consistent with biliary rupture include jaundice, abdominal
distension, and acholic feces.
•
Abdominal pain is most likely with acute rupture or septic peritonitis.
•
Fever may occur with septic peritonitis or cholecystitis.
Laboratory Evaluation
•
Laboratory findings include hyperbilirubinemia and increased ALP, ALT, and SBA concentrations.
•
Abdominal fluid appears yellow or green. Chemical tests for bilirubin are positive
and concentrations of bilirubin are at least 2 times higher in the abdominal fluid
than in the serum.
•
Cytologic examination reveals mixed inflammatory infiltrate and bile-laden macrophages
or free green or yellow-brown material consistent with bile. In some dogs with biliary
rupture, bile pigment is not visible but an acellular amorphous blue mucinous extracellular
material (“white bile”) may be observed. These dogs still have fluid bilirubin concentrations
greater than twice the serum value.
•
Bacteria may be seen if bile peritonitis is complicated by sepsis.
Radiography and Ultrasonography
•
Abdominal radiographs reveal poor abdominal contrast due to fluid accumulation.
•
On ultrasonography, the gallbladder may not be visible, and even a small amount of
abdominal fluid may be detected.
•
Other radiographic and ultrasonographic findings depend on the underlying cause of
rupture, such as gallbladder mucocele, cholecystitis, cholelithiasis, and biliary
neoplasia.
•
When trauma is suspected as the cause of biliary rupture, take thoracic films to detect
other complications such as pneumothorax, diaphragmatic hernia, and bile pleuritis.
Laparotomy
Confirm rupture of the biliary tract by laparotomy.
•
Rupture of the gallbladder secondary to cholecystitis may be acute or chronic. With
chronic gallbladder rupture, omental and hepatic adhesions are common. Biliary fistulas
may develop from the gallbladder to other abdominal structures such as the diaphragm.
Fistulas between the gall bladder and the diaphragm may result in bile pleuritis and
biliary effusion.
•
Submit abdominal fluid and affected biliary tissue for aerobic and anaerobic bacterial
culture.
Treatment
Surgery is required to repair the biliary rupture and is discussed in Chapter 72.
The prognosis is guarded.
Key Point
Dogs with septic peritonitis (positive bacterial cultures of the biliary effusion)
are less likely to survive than are dogs with negative culture results.
•
Prior to surgery, stabilize the patient with fluid therapy, give vitamin K1 parenterally
for 24 to 48 hours, and give antibiotics.
•
Open abdominal drainage is not necessary for dogs with sterile biliary effusions but
may be helpful in managing dogs with septic peritonitis (see Chapter 76).