- Record: found
- Abstract: found
- Article: found
Phylogenetic and morphological analyses of Pilosocereus leucocephalus group s.s. (Cactaceae) reveal new taxonomical implications
Read this article at
Abstract
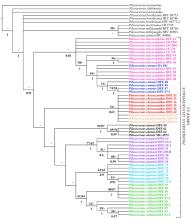
Pilosocereus is one of the Cactaceae family’s most relevant genera in terms of the number of species and its wide geographical range in the Americas. Within Pilosocereus, five informal taxonomic groups have been recognized, one of which is P. leucocephalus group s.s., whose phylogenetic relationships remain unresolved. Therefore, our objectives are to recognize the circumscriptions of the species in P. leucocephalus group s.s. and to corroborate the monophyly and phylogenetic relationships of this group through a set of morphological and molecular characters. This study is based on representative sampling along the broad distribution of this group in Mexico and Central America using multivariate and phylogenetic analyses. The morphological characters identified to contribute to species recognition and group formation are branch diameter, areole length, the areole length-width ratio, the distance between areoles, the length of the longest radial spine, and branch and spines colors. The chloroplast markers rpl16, trnL-trnF, and petL-psbE and the nuclear marker AT1G18270 support the monophyly of the P. leucocephalus group s.s., and two probable synapomorphies are suggested, including one transversion in rpl16 and another in petL-psbE. Together, our results demonstrate that sampled species of P. leucocephalus group s.s. encompass six species distributed in Mexico and Central America: P. alensis and P. purpusii in the western region, P. chrysacanthus and P. collinsii in the central region, and P. gaumeri and P. leucocephalus in the eastern region. A taxonomic key to recognized species is provided.
Related collections
Most cited references58
- Record: found
- Abstract: found
- Article: found
RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies
- Record: found
- Abstract: found
- Article: not found
MUSCLE: multiple sequence alignment with high accuracy and high throughput.
- Record: found
- Abstract: found
- Article: found