- Record: found
- Abstract: found
- Article: found
Familial history and prevalence of BRCA1, BRCA2 and TP53 pathogenic variants in HBOC Brazilian patients from a public healthcare service
Read this article at
Abstract
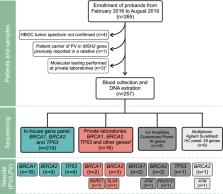
Several studies have demonstrated the cost-effectiveness of genetic testing for surveillance and treatment of carriers of germline pathogenic variants associated with hereditary breast/ovarian cancer syndrome (HBOC). In Brazil, seventy percent of the population is assisted by the public Unified Health System (SUS), where genetic testing is still unavailable. And few studies were performed regarding the prevalence of HBOC pathogenic variants in this context. Here, we estimated the prevalence of germline pathogenic variants in BRCA1, BRCA2 and TP53 genes in Brazilian patients suspected of HBOC and referred to public healthcare service. Predictive power of risk prediction models for detecting mutation carriers was also evaluated. We found that 41 out of 257 tested patients (15.9%) were carriers of pathogenic variants in the analyzed genes. Most frequent pathogenic variant was the founder Brazilian mutation TP53 c.1010G > A (p.Arg337His), adding to the accumulated evidence that supports inclusion of TP53 in routine testing of Brazilian HBOC patients. Surprisingly, BRCA1 c.5266dupC (p.Gln1756fs), a frequently reported pathogenic variant in Brazilian HBOC patients, was not observed. Regarding the use of predictive models, we found that familial history of cancer might be used to improve selection or prioritization of patients for genetic testing, especially in a context of limited resources.
Related collections
Most cited references83

- Record: found
- Abstract: found
- Article: found
Fast and accurate short read alignment with Burrows–Wheeler transform

- Record: found
- Abstract: found
- Article: found