
INTRODUCTION
Annual seasonal circulations of influenza virus cause diseases in people that can account for significant mortality – from 290,000 to 650,000 people – in the world population according to the estimates of the World Health Organization (WHO) [1, 2]. There are four known types of the influenza virus – A, B, C, and D, of which only type A and B viruses cause seasonal epidemics. Influenza A viruses in turn are divided into subtypes depending on the type of the main surface glycoproteins of the virus – hemagglutinin (HA) and neuraminidase (NA). Currently, 18 subtypes of HA (HA1 HA18) and 11 of NA (N1 N11) are known. The two subtypes – A(H1N1) and A(H3N2) – are responsible for the seasonal influenza epidemics. For influenza B virus, two main lineages B/Yamagata and B/ Victoria are known. A broad variety of existing antigenic variants of the influenza virus is caused by its rapid evolutionary variability [2, 3]. The high frequency of mutations that occur in the genome of the influenza virus, represented by negative-sense single-stranded segmented RNA, and the lack of proofreading activity of the viral RNA polymerase lead to the emergence of a large number of mutations that eventually cause the formation of new antigenic variants. This phenomenon is known as antigenic drift. The segmented structure of the influenza virus genome determines the ability to exchange fragments between the strains of the same virus type, which leads to the emergence of new viruses and is called antigenic shift. The survival of viable viral mutants is controlled by natural selection.
The main licensed drugs that are currently used for the prevention and treatment of influenza A and influenza B infections are the NA inhibitors – zanamivir, oseltamivir, laninamivir, and peramivir [4]. The mechanism of action of these drugs is based on blocking the function of viral NA and that leads to the limited spreading of the virus in the respiratory tract [5]. Peramivir and laninamivir inhibit NA activation for a much longer period of time than oseltamivir or zanamivir [6, 7]. However, the treatment of patients with these drugs causes mutations in the NA gene and that leads to the formation and spread of drug resistant viruses.
The analysis of the nucleotide sequences of HA and NA genes of the viruses isolated from patients in different parts of the globe is widely used to predict the evolutionary variability of the influenza virus as well as to predict the emergence of new epidemic strains using bioinformatics methods, in particular by phylogenetic analysis [8]. In recent years, phylogenetic analysis has become one of the main components of epidemiological surveillance, which is used to determine the relationship and evolution of influenza viruses [9]. Despite the fact that many research projects devoted to the study of the selection pressure on the genes of human influenza viruses have already been performed, the relationship between positively selected sites, antigenic variability of the virus, and sensitivity to NA inhibitors are still not fully understood [10-12].
The goal of the present study was to explore the effect of selection pressure on the NA gene of the influenza viruses isolated in Ukraine in 2009-2015 as well as the identification of specific mutations in the NA gene of influenza virus isolates associated with resistance to NA inhibitors.
The most common approach used to determine the selection pressure on proteins is the comparison of the nonsynonymous (dN) and synonymous (dS) substitution rates at the sites that are undergoing selection [13, 14]. The dN/dS ratio quantifies the selection pressure by comparing the rate of these substitutions. If natural selection contributes to the fixing of the changes in the protein sequence, the value of the dN/dS ratio is greater than 1, while if natural selection inhibits changes in the protein, the value of this ratio is less than 1. If the value of the dN/dS ratio is close to 1, the selection pressure is absent. We applied this method to study the selection pressure on the NA gene of human influenza A viruses of A(H1N1)pdm09 and A(H3N2) subtypes as well as influenza viruses of type B of both main lineages B/Yamagata and B/Victoria isolated in Ukraine in 2009-2015. The study of the effect of selection pressure on the HA gene was not a part of this project.
MATERIALS AND METHODS
Viruses
The influenza viruses isolated in epidemic seasons 2009-2015 were used in this study. Samples were obtained from patients with influenza-like illness (ILI) and severe acute respiratory infections (SARI) in the form of nasal swabs, nasopharynx, or oropharynx. In addition, influenza virus isolates obtained from the sentinel centers in Kiev, Odessa, Dnipro, and Khmelnitsky were also included in this study. The diagnostics of SARI and ILI in patients was accomplished according to the WHO criteria [15].
Cell lines
The cell line of Madin-Darby canine kidney (MDCK), which was used in this study, was obtained from the Smorodintsev Research Institute of Influenza (St. Petersburg, Russian Federation). Influenza A(H3N2) viruses were isolated on a genetically modified MDCK-SIAT1 cells obtained from the Centers for Disease Control and Prevention (CDC, Atlanta, USA) [16].
Real-time RT-PCR
Isolation of RNA from the influenza viruses was carried out using the QIAamp® Viral RNA Mini Kit (QIAGEN, Germany) according to the manufacturer’s instructions. The reaction mixture for PCR was prepared according to the method for the detection of swine and seasonal A(H1N1)pdm09 influenza viruses as recommended by the CDC (Atlanta, USA) [17]. For the PCR, the Ambion AgPath-IDTM One-Step RT-PCR Kit and a set of primers and probes (Thermo Fisher Scientific, USA) were used. The reaction was carried out in a real-time PCR system Applied Biosystems real-time PCR 7500 (Thermo Fisher Scientific, USA).
Gene sequencing of influenza viruses
Gene sequencing of the isolated influenza A viruses of A(H3N2) and A(H1N1)pdm09 subtypes and type B influenza viruses was carried out at the WHO Influenza Center in London (National Institute for Medical Research, NIMR, London, UK) and at the CDC (Atlanta, USA) according to the standard protocol [18]. All of the obtained nucleotide sequences were deposited to the international database – Global Initiative on Sharing Avian Influenza Data (GISAID, http://platform.gisaid.org/).
Phylogenetic analysis
The sequences of the NA genes obtained from influenza viruses isolated in the 2009-2015 epidemic seasons were used in this study. A total of 375 Ukrainian influenza virus isolates were examined. Of these, 142 sequences belong to influenza A type A(H1N1)pdm09 subtype, 110 sequences belong to influenza A(H3N2) subtype, 123 sequences – to type B influenza viruses (94 sequences to the B/Yamagata genetic lineage and 29 sequences to the B/Victoria genetic lineage). For the identification of mutations, the comparison of the obtained sequences was carried out using BLAST analysis (https://blast.ncbi.nlm.nih.gov/Blast.cgi). For phylogenetic analysis, we selected Ukrainian and European isolates obtained in the course of the study period as well as vaccine and reference strains. Multiple sequencing alignment was performed using the ClustalW software [19]. Phylogenetic analysis was performed by means of the ML (maximum likelihood) method using the Tamura 92 (T92) model with an approximation of individual site frequencies by gamma distribution with bootstrap 1,000 replications. The construction of phylogenetic trees was carried out with MEGA6 software [20, 21]. The final trees were visualized using FigTree 1.4.2 software [22].
The amino acid substitutions leading to resistance to NA inhibitors were identified: E119I (V/D), Q136K, D151E/V I222L, R224K, E276D, R292K, N294S, R371K – for influenza A viruses of A(H3N2) subtype; I117V, E119G/V, Q136K, Y155H, D198G, I222K/R/V, S246N/G, H274Y, N294S – for viruses of A(H1N1)pdm09 subtype; E119A (G, V, A/D), R152K, D198E (N/Y), I222T (V/I), H274Y, R292K, N294S, R371K, G402S – for type B influenza viruses. Numbering was carried out according to N2 [23].
Study of selection pressure
The study of the NA gene sequences of influenza viruses was carried out using the Datamonkey server [24, 25] and the following methods: SLAC (single-likelihood ancestor), FEL (fixed-effects likelihood), IFEL (internal branch fixed-effects likelihood), included in the software package HyPhy of the Datamonkey server. Furthermore, the method FUBAR (fast unconstrained Bayesian approximation) and MEME (mixed-effects model of evolution) were used. The ratio dN/dS was determined using HyPhy.
RESULTS
Influenza A(H1N1)pdm09 viruses
One site (40) that is under positive selection pressure was detected in NA protein of the influenza A(H1N1) pdm09 viruses: the L40V mutation was found in four isolates from the 2012-2013 season, and L40I mutation in viruses circulating in the 2013-2014 and 2014-2015 seasons (Table 1, Fig. 1). A modest effect of positive selection pressure on pandemic influenza viruses was also recorded by other authors, although they revealed mutations at different sites [12].
| Type/subtype of the virus | dN/dS | Sites under the selection pressure | ||||
|---|---|---|---|---|---|---|
| positive | negative | |||||
| Site | Mutation | Season, years | Total number of sites | Sites, studied by 4 methods * | ||
| A(H1N1)pdm | 0.261 | 40 | L40V | 2012-2013 | 27 | 2 sites: 395, 439 |
| L40V | 2013-2015 | |||||
| A(H3N2) | 0.205 | 93 | D93E | 2010-2011 | 58 | 10 sites: 106, 107, 128, 182, 296, 306, 364, 427, 432, 449 |
| D93G | 2011-2012 | |||||
| 402 | N402D | 2010-2011 | ||||
| B/Yamagata | 0.226 | 74 | L74P | 2014-2015 | 52 | 7 sites: 64, 83, 114, 173, 272, 329, 428 L83P |
| 99 | S99I | |||||
| 268 | T268K | |||||
| B/Victoria | 0.292 | 358 | A358K | 2011-2012 | 17 | 1 site: 371 |
| 288 | E288Q | 2010-2011 | ||||
| 455 | L455I | |||||
Sites were studied by SLAC, FEL, IFEL, and FUBAR methods
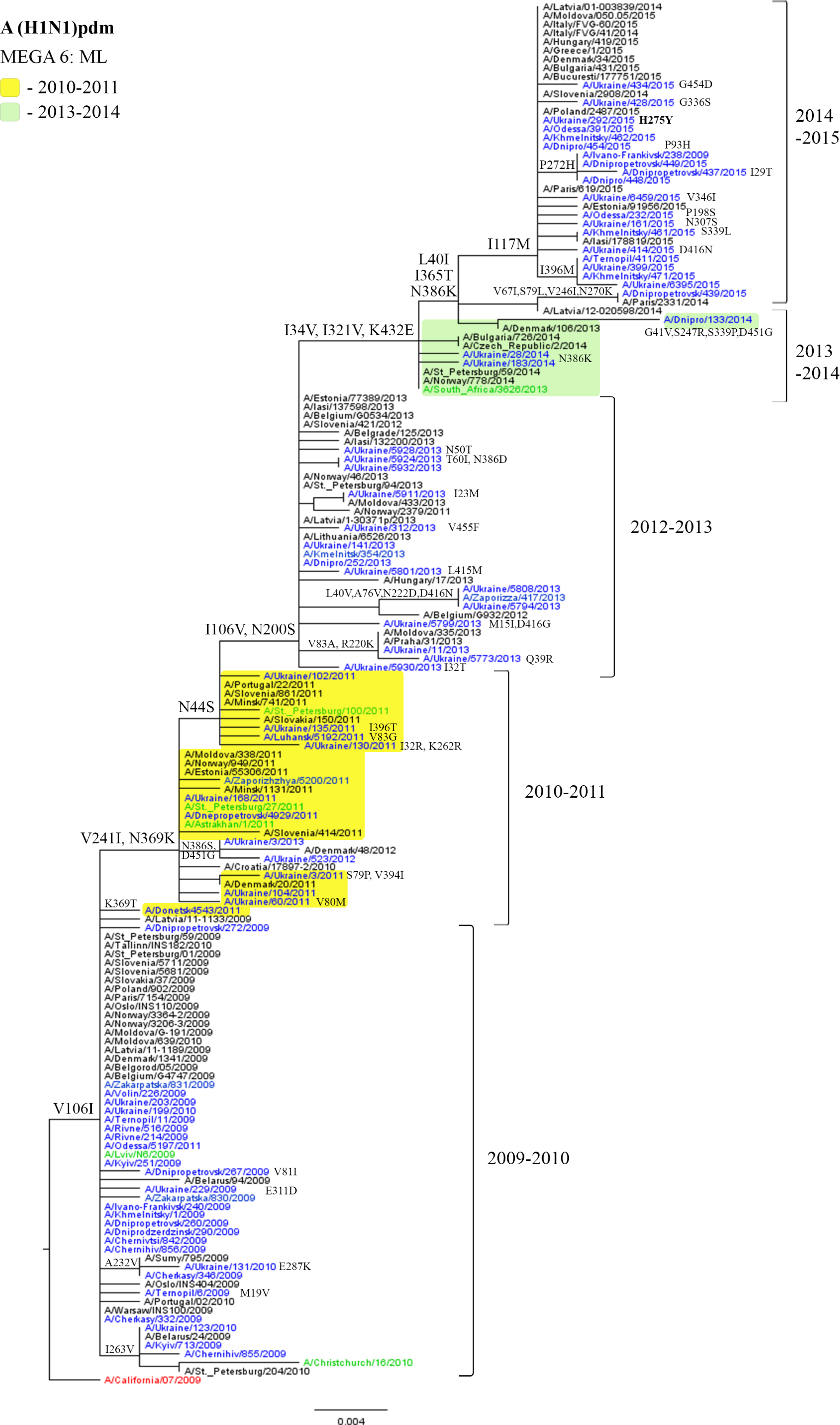
Viruses isolated in Ukraine are marked in blue (viruses of the 2010-2011 season are highlighted in yellow, viruses isolated in 2013-2014 are highlighted in light green); the vaccine strain is marked in red; the reference strains are marked in green.
Phylogenetic analysis of influenza viruses of the A(H1N1)pdm09 subtype revealed a high level of identity, 99%. All Ukrainian influenza viruses were similar to the A/California/07/2009 virus that was used for the vaccine strain preparation.
Although Ukrainian isolates of the A(H1N1)pdm09 subtype acquired unique mutations, the majority of substitutions were located in the same sites as in the isolates from the neighboring countries [26–31]. For example, all the Ukrainian isolates of the 2014-2015 epidemic season contained the typical I117M substitution and were similar to the strains from European countries such as Italy, Latvia, Poland, Slovakia, France, Estonia, etc. Similar viruses were isolated in New Zealand [32]. These results suggest that mutations in the NA gene did not lead to significant changes of the NA antigenic properties of the influenza viruses of this subtype.
Influenza A(H3N2) viruses
Two sites that are under positive selection pressure – 93 and 402 – were detected in influenza viruses of A(H3N2) subtype using at least one of the following methods – FEL, IFEL, and FUBAR (Table 1).
The Ukrainian isolate A/Ukraine/175/2011 acquired mutations D93E and D402N and was similar to the isolates from Belarus and Estonia as well as to the Ukrainian isolates from the next epidemic season. D93E mutation was observed only in one isolate A/Ukraine/5381/2012 in the next season. However, in the 2012-2013 season, a group of viruses with D93G mutation was isolated again. Therefore, the D93E mutation appeared in the population of Ukrainian influenza viruses in the 2010-2011 season, and in the next season (2011-2012) amino acid 93G was observed at this position, and the D93G mutation was still present in the population of Ukrainian viruses in the 2012-2013 season (Fig. 2). Belanov et al. consider the D93G mutation as one of the evolutionary markers of influenza viruses of A(H3N2) subtype [33]. Starting from the 2012-2013 season, the virus used for the preparation of the vaccine strain also contained this mutation.
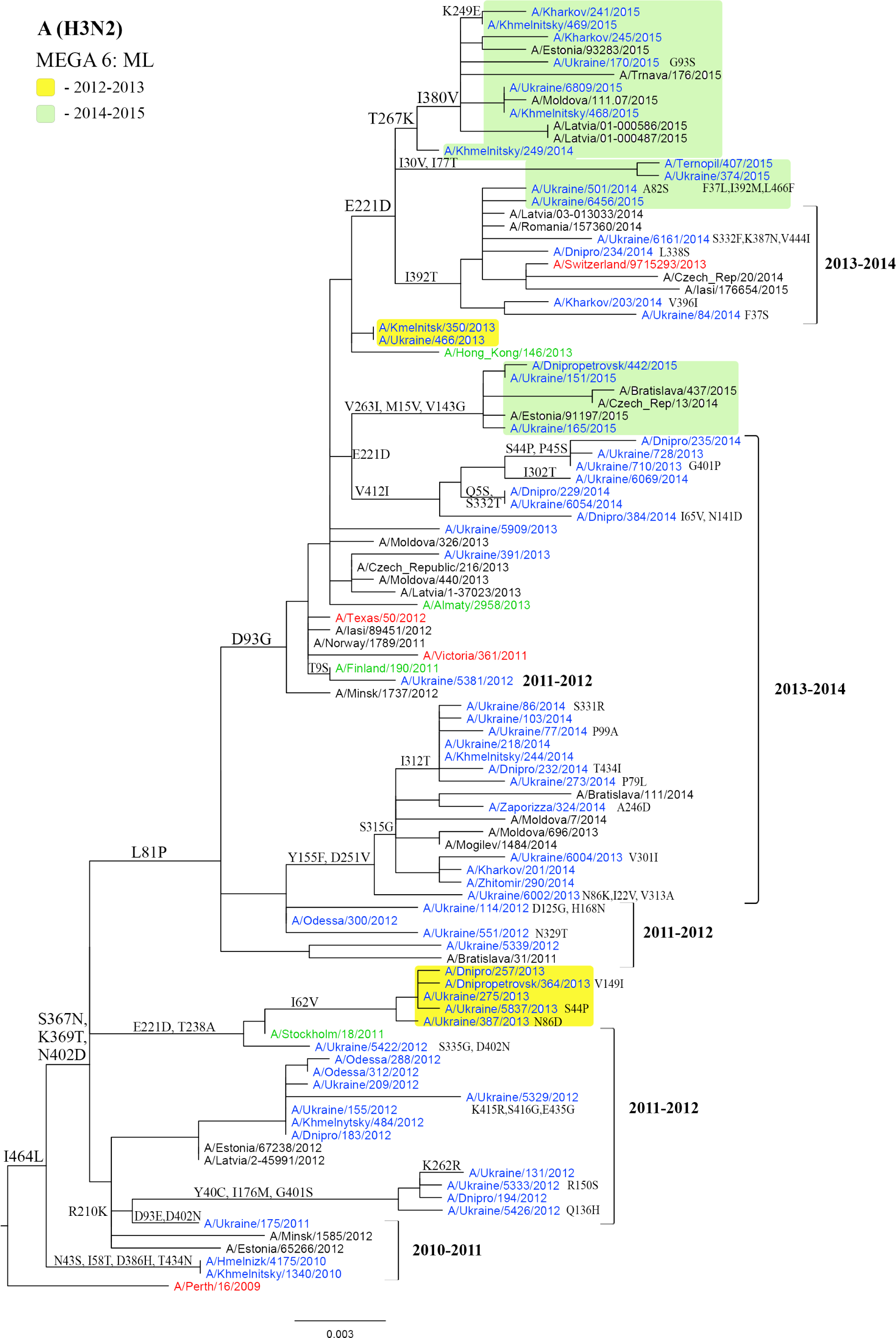
Viruses isolated in Ukraine are marked in blue (viruses of the 2010-2011 season are highlighted in yellow, viruses of 2013-2014 are highlighted in light green); the vaccine strains are marked in red; the reference strains are marked in green.
The analysis of the sequences of viruses from accessible databases (Influenza Virus Resource (IVR) and GISAID) revealed substitution T267K that is considered to be an evolutionary marker, which was observed in some Ukrainian isolates from the 2014-2015 season.
Phylogenetic analysis of influenza A(H3N2) viruses revealed a high level of genetic identity of these viruses, 98.3%.
It is noteworthy that in the 2013-2014 season, the two viruses A/Ukraine/710/2013 and A/Ukraine/728/2013, which only differed by one substitution G401D in the NA protein, were isolated from the same person within a few days. The amino acid at this position is the part of the NA antigenic site [34].
Type B influenza viruses of the B/Yamagata lineage
The study of influenza B viruses of the B/Yamagata genetic lineage showed the presence of three sites – 74, 99, and 268 – under the positive selection pressure (Table 1). Substitutions T268K, S99I, and L74P were observed only in the isolates of 2014-2015 season. At the same time, the isolates with L74P mutation form a separate group on a phylogenetic tree (Fig. 3).
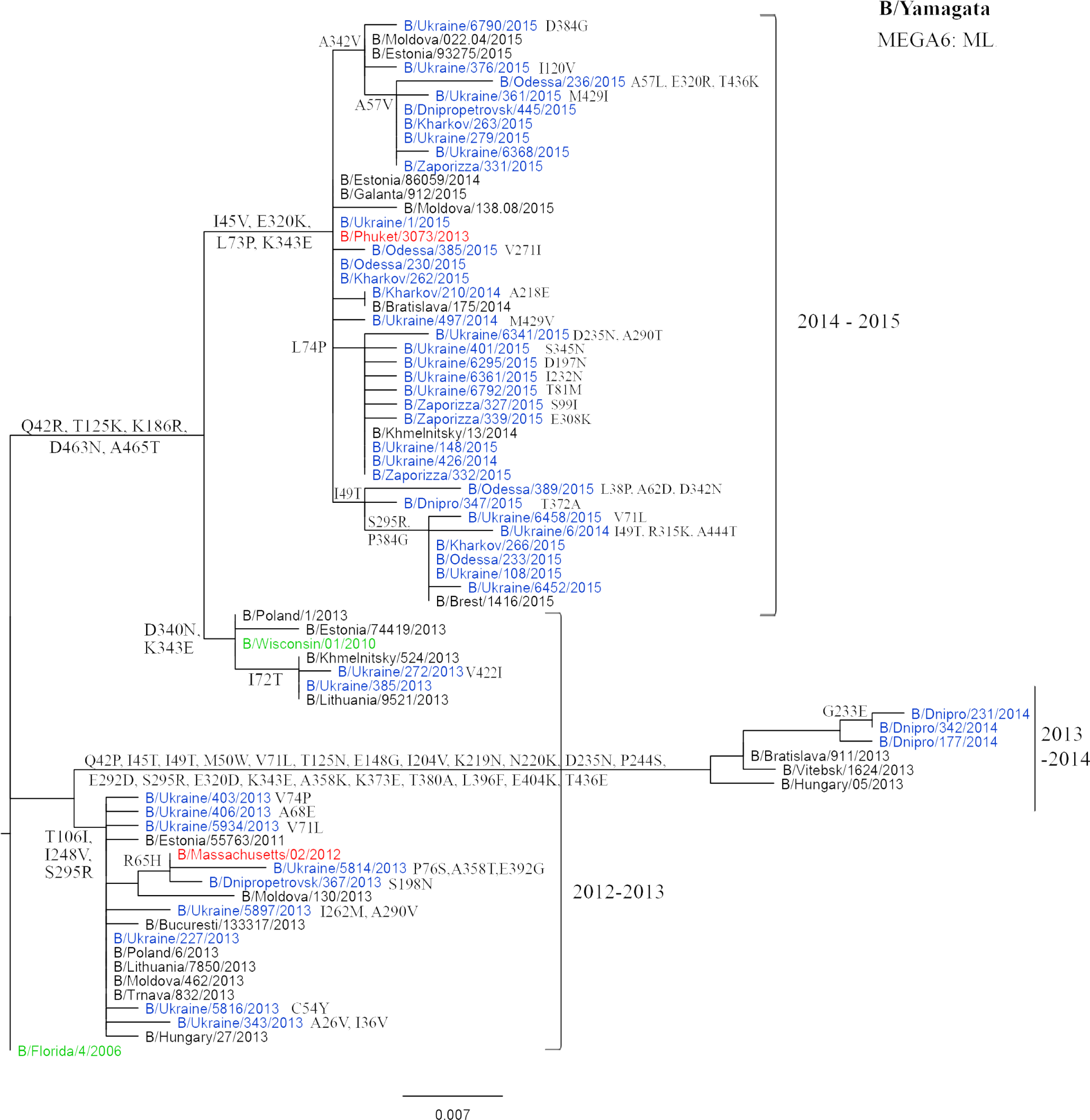
Viruses isolated in Ukraine are marked in blue; the vaccine strains are marked in red; the reference strains are marked in green.
Phylogenetic analysis of the NA gene of influenza B/Yamagata viruses during the studied period showed high genetic identity, 97.5%. Influenza viruses isolated in Ukraine during the studied period were similar to the viruses recommended for the preparation of vaccine strains in the same epidemic season.
Some of the found substitutions theoretically can affect the conformation of the NA protein. Some Ukrainian isolates contained substitutions at potential glycosylation sites (A465T, R65H) or NA stem region (R65H). In addition, substitutions at the T125K and V71L (loop-120) were found. The amino acid substitutions T125K, E148G (loop-150), and D235N (helix-190) were found in some viruses isolated in the 2013-2014 season [26]. According to the data of the Influenza Center in London [27], the influenza viruses isolated in this season contained individual substitutions that were atypical for the Ukrainian isolates, namely: T46I, A55T, G70R, E77A, I262T, R295H, A358T, and K382R.
Type B influenza viruses of the B/Victoria lineage
For influenza viruses of type B of the genetic lineage B/ Victoria, the following sites under the positive selection pressure were found: 358, 288, and 455 (IFEL, MEME) (Table 1). In some cases, only the single mutations E288Q and L455I were found in Ukrainian isolates circulated during the 2010-2011 epidemic season (Fig. 4). However, the A358K mutation, which appeared in viruses circulated in the 2011-2012 season, was observed in all the isolates of the B/Victoria lineage. This result shows that the above-mentioned substitution has been fixed in the influenza virus population as a result of positive selection pressure. A phylogenetic comparison of the NA genes of the B/Victoria lineage isolates revealed a high level of genetic identity of 98.4%.

DISCUSSION
The gradual accumulation of mutations in the gene leads to a change in the structure of the encoded protein, which in turn can lead to a change in the biological properties of the virus, such as the rate of virus spreading, the severity of the disease caused by the virus, contagiousness, sensitivity to drugs, etc. [2]. The study of mechanisms of molecular adaptation of influenza viruses is one of the fundamental goals of evolutionary biology. Nonsynonymous mutations are fixed in the population more often than synonymous ones. This is usually observed when new mutations provide certain advantages for the protein (virus); then, they have a greater chance of fixation. This phenomenon is regularly observed in the course of the evolution of RNA-containing viruses leading to changes of surface antigens HA and NA in the case of influenza virus.
The present paper presents the results of the first study of the selection pressure effect on the variability of the NA protein of human influenza viruses and the emergence of resistance to NA inhibitors conducted in Ukraine. The analysis results of the NA sequences of viruses isolated in Ukraine in the period from 2009 to 2015 are as follows: the dN/dS ratio was 0.261 for NA of the influenza A viruses of A(H1N1)pdm09 subtype, 0.205 for NA of viruses of A(H3N2) subtype, 0.226 for NA of type B viruses of B/Yamagata lineage, and 0.292 for NA of B/Victoria lineage viruses. The obtained data show that the number of synonymous substitutions prevails over the number of nonsynonymous ones for each site of NA protein. This means that for most of the substitutions in the NA gene, the selection pressure is absent.
The values of the dN/dS ratio determined in this study for the NA of the influenza viruses of different types and subtypes are in good agreement with the data reported by other authors. Therefore, Mostafa et al. reported a dN/ dS ratio equal to 0.22 for the NA of the influenza A(H3N2) subtype viruses isolated in Germany in 2015 [35], and a similar value for this ratio was reported by Li et al. who studied 1,397 viruses of the A(H1N1)pdm09 subtype in China [12]. In the course of analysis of 98 sequences of NA of the influenza B type viruses of B/Yamagata lineage by Horthongkham et al. in Bangkok [36], the dN/dS ratio was found to be 0.20.
We found that only some sites in the NA turned out to be under positive selection pressure: site 40 for viruses of A(H1N1)pdm09 subtype, sites 93 and 402 for influenza A(H3N2) viruses, sites 74, 99, and 268 for influenza B viruses of the B/Yamagata lineage, and sites 358, 288, and 455 for the viruses of the B/Victoria lineage. These sites do not belong to the active center, transmembrane domain, or antigenic sites of NA. However, as a result of the selection pressure, mutations at these positions were fixed in the influenza virus population and these viruses often formed a separate branch on the phylogenetic tree. These results were proved by analyzing a number of selected sites by all four methods (SLAC, IFEL, FEL, and FUBAR). In one of the isolates of the B/Yamagata lineage – B/Odessa /389/2015 – the L83P mutation was detected. Since this mutation was not found in the influenza virus population in the 2017-2018 season, it was not a fixed mutation.
The treatment of influenza with NA inhibitors can create selective pressure on the emergence of virus resistance to these drugs, which can in turn affect the genetic diversity of the viral population. The emergence of the viral resistance to the antiviral drugs is an important problem of public health care. However, in the course of our study, no positive selection pressure was found on the sites associated with resistance to NA inhibitors. Therefore, in Ukraine, NA inhibitors are not a significant factor in the process of selection of the influenza viruses. Apparently, this is due to the fact that NA inhibitors are not widely used for the treatment of influenza infections in Ukraine. However, these studies should be continued.