
INTRODUCTION
Cancer is the second leading cause of death worldwide. According to the World Health Organization (WHO), 9.9 million deaths from cancer were registered worldwide in 2020 [1]. In the same year, 556,036 cases of oncological diseases were detected in Russia as reported by the State Cancer Registry. According to the statistics, the most common localizations in the overall cancer incidence in Russia are the mammary gland (11.8%), skin (10.9%), trachea, bronchi, lung (9.8%), colon (7.2%), prostate gland (7.9%), stomach (5.8%), rectum, rectosigmoid junction (5.1%), lymphatic and hematopoietic systems (5%), uterine body (4.3%), kidney (3.8%), pancreas (3.4%), cervix (2.8%), urinary bladder (2.8%), and ovary (2.4%) [2].
Until recently, the main methods of cancer treatment have been chemotherapy and radiation therapy, which have serious side effects. Cancer drug therapy is less painful and can be the preferred treatment in many cases. However, many tumors are resistant to the standard treatments, which leads to relapses of disease over time. One of the new approaches for cancer treatment is oncolytic virotherapy, which is actively developing in the last decade. The most difficult problem in oncolytic virotherapy is the choice of a virus that can infect a tumor, destroy it, and induce an immune response to tumor cells. Antitumor efficacy in animal experiments has been shown for many viral families, which has led to the initiation of clinical trials for the treatment of various forms of cancer.
Oncolytic virotherapy has a long history. Back in 1904, George Dock described a clinical case with a sharp decrease in the number of leukocytes in a patient with leukemia after an influenza infection [3]. In the same year, the Italian physician Nicola De Pace described the disappearance of a malignant tumor of the cervix in a patient after immunization with a live rabies vaccine due to a dog bite. Immunization of several similarly diagnosed patients with attenuated rabies vaccine led to a decrease in the tumor size, but later the disease recurred with a lethal outcome in all patients [4]. In 1940, Pack conducted the first clinical trial using a rabies vaccine to treat melanoma which caused partial remission [5]. In the 1960s, a study by Lindenmann and Klein showed that immunization of mice with a homogenate of tumor cells infected with the influenza virus led to a humoral immune response against the antigens associated with tumor cells [6, 7].
Wild-type viral strains that are non-pathogenic or low pathogenic for humans can serve as oncolytic viruses without genetic modifications (for example, reovirus) [8]. In addition, genetically modified viruses with a decreased pathogenicity and increased therapeutic effect are designed [9, 10]. The development of recombinant DNA technologies has created new possibilities to obtain a wide range of chimeric (recombinant) viruses with modifications aimed at destroying cancer cells. The first of such studies was published in 1991 and described a genetically modified herpes simplex virus type 1 (HSV1, dlsptk) with a reduced neurovirulence due to the removal of the γ 34.5 gene that encodes thymidine kinase (HSV-TK) [11]. The modification of the viral genome reduced the reproduction of the virus in normal cells while retaining normal reproduction in tumor cells containing an increased concentration of TK [12]. The dependence of the TK-defective virus on the level of this enzyme in the cell allowed for using the modified virus for the treatment of glioma in mice, subsequently leading to tumor regression.
To date, nine viral families with oncolytic potential have been described, namely: Rhabdoviridae, Poxviridae, Herpesviridae, Reoviridae, Adenoviridae, Paramyxoviridae, Picornaviridae, Togaviridae, and Parvoviridae. The representatives of these families have shown a positive therapeutic effect in the treatment of malignant neoplasms of various origins and localizations. Several oncolytics have been developed and approved for use in clinical practice, including a genetically modified enterovirus ECHO-7 (Latvia) named RIGVIR® which has oncolytic and immunomodulatory properties and is adapted to melanoma [13]. In 2003, the China Food and Drug Administration (CFDA) approved a new medication Gendicine® based on the adenoviral (Ad) vector with E1-deletion for the treatment of head and neck cancer. However, the replication-deficient oncolytic virus had low transfection efficiency for cancer cells. Nevertheless, Gendicine® is a commercially available medication that is used in clinical practice. The investigation of this drug for the treatment of lung cancer, liver cancer, gynecological malignancies, and soft tissue sarcomas is still ongoing [14]. The oncolytic based on a genetically modified adenovirus H101, named Oncorine®, was approved in China in 2005 for the treatment of malignant head and neck tumors originating from squamous epithelial cells [15]. The FDA and the European Medicines Agency (EMA) have approved the HSV1-based anticancer oncolytic Talimogene laherparepvec (T-VEC®) for the treatment of inoperable dermal, subcutaneous, and nodular lesions in patients with melanoma recurrence after surgical removal of the primary tumor [16, 17]. This virus is modified for selective growth in tumor cells and expression of granulocytemacrophage colony-stimulating factor (GM-CSF) that has immunostimulatory activity.
Thus, oncolytic viruses are promising antitumor agents that can be used both for independent therapy of malignant neoplasms and for treatment in combination with surgical methods, chemotherapy, radiation therapy, or immunotherapy.
Main mechanisms of action of the oncolytic viruses
According to the current classification, eight main hall-marks distinguish tumor cells from healthy ones, which include: maintenance of proliferative signaling, evasion of growth suppressors, resistance to cell death, enabling replicative immortality, induction of angiogenesis, activation of invasion and metastasis, reprogramming of energy metabolism and evasion of immune destruction [18, 19]. The possibility to direct viruses to various cellular targets determines the strategy for choosing a virus for the treatment of human cancer.
The effectiveness of oncolysis depends primarily on the ability of viruses to infect and destroy cancer cells selectively. Some wild-type viruses such as vesicular stomatitis virus (VSV), HSV, Newcastle disease virus (NDV), and reovirus predominantly infect tumor cells, reproduce using the cell’s replication machinery [20-23], and cause cancer cell death by apoptosis, necrosis, or autophagy [24-27]. Other viruses, such as adenovirus, require modification of the viral genome to limit replication in normal cells. This mechanism is called targeting. Thus, the p53 protein – a transcription factor that blocks the cell cycle and induces apoptosis – is not functional in most tumor cells due to direct mutations in the p53 or in proteins that control its activity or stability like p14ARF and Mdm2 [28, 29]. The early adenovirus gene E1B encodes the E1B 55K protein, which binds to p53 and protects the infected cell from premature death. Therefore, the adenovirus with the non-functional E1B 55K protein replicates in tumor cells, whereas in normal cells its replication is abortive [30-33]. Other DNA viruses, such as SV40 or human papilloma virus 16 (HPV16), are also able to deactivate the p53 protein [29].
Another example is the influenza A virus with the de-letion in the gene encoding the nonstructural NS1 protein (delNS1) that promotes virus resistance to the host interferon (IFN) system. This modified virus is able to kill cancer cells carrying a defective P53 gene via an alternative p53-independent pathway [34, 35]. The ability of mammalian cells to secrete type I IFNs is an important innate immunity mechanism against viral infections. Tumor cells may have defects in the genes that are involved in the synthesis of IFNs, resulting in a decreased expression of these genes [36-40]. The absence of a functioning IFN system in cancer cells makes them more sensitive to infection with cytolytic viruses [35-38]. Thus, the delNS1 influenza virus has an increased cytolytic potential in cancer cells [39] and reduced reproductive activity in healthy cells with an active IFN system [40, 41]. Such influenza viruses can serve as directed killers of cancer cells. Similarly, the representative of another viral family HSV1 fights the host’s IFN system with the ICP0 protein (expressed by the γ 34.5 gene). Deletion of this gene in the HSV1 genome enables active replication of the virus in cancer cells selectively, but not in healthy cells with an active IFN system [42].
The therapeutic effect of viruses is determined not only by the lysis of cancer cells but also by stimulation of antitumor immunity and elimination of immune suppression through pro-inflammatory activation of the tumor immune microenvironment (TIME) [43-49]. Cancer cells can escape the immune system by means of mutations in tumor antigens. By infecting tumor cells, oncolytic viruses trigger an inflammatory response that promotes the recognition of virus-infected cancer cells by host immune cells. This results in the induction of cytokines that activate the immune system, leading to the recruitment and activation of dendritic cells (DCs), stimulation of specific T lymphocytes, and activation of an effective antitumor response. During apoptosis and/or necrosis, damage-associated molecular patterns (DAMPs) are released [50]. They are recognized by the antigen-presenting T cells in the tumor microenvironment, which eventually leads to tumor death.
In order to enhance the cytolytic immune effect, oncolytic viruses are engineered to express additional immunostimulatory factors. This phenomenon is known as arming. Cytokines, chemokines, costimulatory proteins, bispecific T cell activators (BiTE), or immune checkpoint blockers are used as transgenes. For example, the oncolytic virus JX-594 of the Poxviridae family is the modified smallpox vaccinia virus (VV) strain Wyeth, in which the gene encoding TK has been removed to reduce the virulence and limit the reproduction in normal cells, while the gene expressing the immunostimulatory GM-CSF protein and the lacZ reporter gene has been inserted in order to activate antigen-presenting cells [51-53]. Another example is adenovirus LOAd703 (serotype 5/35), an oncolytic virus for the treatment of patients with pancreatic cancer, that expresses the human CD40L (TMZ-CD40L) and 41BB (4-1BBL) ligands activating the CD40 and 4-1BB pathways [54]. This genetic combination enhances the Th1 immunity by increasing the production of cytokines and co-stimulatory molecules. Adenoviruses can generate cytotoxic proteins while replicating. For instance, the adenovirus death protein (ADP) synthesized in the late stage of infection causes cell lysis. During lysis, new virions that infect other tumor cells are released [55, 56]. The modified human adenovirus C serotype 5 (Ad5), which contains both ADP and a chimeric suicide gene or sodium-iodide symporter (NIS, a human reporter gene), was shown to selectively suppress the growth of prostate tumor cells [57-59]. Ad5 adenoviruses are widely used to create new oncolytic viruses. However, the disadvantage of Ad5 is its wide distribution in the human population and, as a result, the formation of immune resistance in up to 90% of the population [60, 61]. The pre-existing immunity to adenovirus may limit the infection of cancer cells with the corresponding oncolytic virus, as well as increase the viral toxicity to liver cells and the blood coagulation system [62, 64, 65]. It is necessary to increase the specificity of the oncolytic virus to the cancer cells to overcome the immune resistance to oncolytic virotherapy. For this purpose, special elements regulating the specific replication of the virus in the cancer cell are included in the viral genome, which reduces the overall viral toxicity and promotes a stronger immune response to the tumor [62, 64, 65]. Another approach to overcome the immune barrier is to use new, less common oncolytic adenoviruses of other serotypes for the genome construction, e.g., Ad26 or Ad24 [61, 63]. The possibility of using type B adenoviruses (Ad11p), to which the majority of the population is not immune, is also being studied [66].
In order to overcome the immune barrier, it is also possible to sequentially apply several oncolytic viruses that do not have cross-immunity. The sequential virotherapy has an advantage due to the ability of cytolytic viruses of various families to trigger different mechanisms of programmed death (apoptosis, pyroptosis, autophagic cell death, necroptosis, etc.) and inflammation of cancer cells [27]. This approach allows for creating a platform for a polyvalent effect focused on a deeper damage and eradication of the tumor and its metastases, affecting chemotherapy-resistant stem cancer cells as well. The diagram that illustrates the principle of sequential virotherapy using oncolytic viruses of various families is presented in Fig. 1.
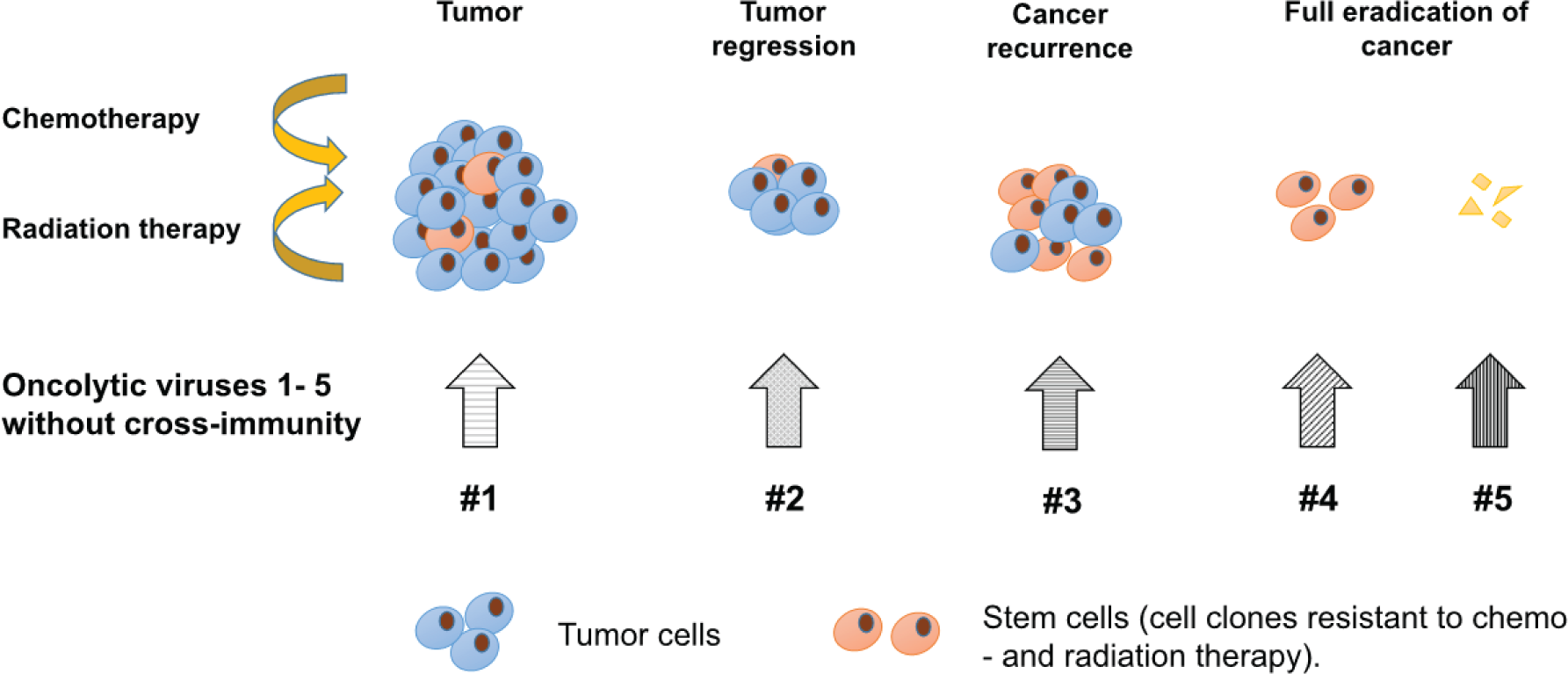
Sequential virotherapy allows: (1) to avoid the immune reaction of the macroorganism that neutralizes the oncolytic virus; (2) to modify the virus and at the same time adjust the therapy, depending on the type and location of the tumor (or its metastases); (3) to selectively induce the death of cancer cells through the apoptotic and/ or necrotic pathway, depending on the properties of the oncolytic virus, and thereby affect the level of the inflammatory response in the tumor; (4) to develop specifically optimized sets of viruses for the treatment of tumors of various origins to achieve their complete eradication in the future. As a result, the complete eradication of both the primary tumor cells and their stem clones becomes possible.
In addition to the modifications of viruses known as targeting and arming, another modification of oncolytic viruses – shielding – is used to increase the virus spread. For example, if a patient is immune to an oncolytic virus, biodegradable synthetic polymers can be used to deliver the virus in order to prevent its neutralization. Currently, several polymers, e.g., a cationic polymer associated with polyethylene glycol (PEG), poly-(N-(2-hydroxypropyl) methacrylamide) (pHPMA) [67, 68], and a DNA aptamer, are being studied for this purpose [69].
The blood circulation in the tumor, which supports the growth and metastasis of cancer cells, can be damaged by oncolytic viruses. The formation of new blood vessels (angiogenesis) is controlled by chemical signals, such as vascular endothelial growth factor (VEGF) that binds to receptors on the surface of normal endothelial cells. This triggers signaling pathways in endothelial cells, which promote the formation and growth of new blood vessels. Angiogenesis inhibitors interact with VEGF, preventing its activation and formation of new blood vessels. Virus-induced suppression of angiogenesis in malignant tumors has been shown for adenovirus [70, 71], herpesvirus [72], VV [73], vesicular stomatitis virus (VSV) [74, 75], and measles virus [76, 77]. To increase the efficiency of virus penetration into the tumor vasculature, the viruses can be targeted to vascular endothelial cells. An example is the non-replicating adenovirus vector Ad5 with an E1 deletion modified with a mouse promoter (PPE-1-3X), Fas transgene, and tumor necrosis factor receptor 1 (TNFR1). This vector, named VB-111, showed an antitumor effect in clinical trials by infecting the angiogenic vasculature [70]. The antitumor effect has also been shown for the adenovirus FGF2-Ad-TK expressing HSV-TK, which targets fibroblast growth factor 2 (FGF2) [71]. In HSV1 oncolytic vectors, the γ 34.5 gene is deleted to reduce the neurovirulence, leading to efficient selective virus replication only in dividing cells [72]. Antiangiogenic HSV1 oncolytic vectors express angiogenesis inhibitors or target pro-angiogenic factors [78]. The modified VV (JX-594), which selectively targets the tumor vasculature by activation of the ras/MAPK signaling pathway, has been shown to cause the collapse of tumor vessels when administered intravenously [73]. The JX-594 virus, which expresses the hGM-CSF transgenes and β-galactosidase, replicates in the endothelial cells of the tumor vessels in mice when injected intravenously, causing tumor necrosis. Effective suppression of the tumor growth was also observed when the mutant VSV (VSVDelta51) was used in combination with the vascular disruptor agent ZD6126 [75].
One of the problems in treating cancer is its recurrence due to the presence of tumor cells that are resistant to the traditional treatment methods [79]. Regarding their properties, these cells have a lot in common with normal stem cells; therefore, they are called cancer stem cells (CSC) [80]. These cells can self-renew, they are pluripotent and can remain dormant without dividing for a long time [81]. CSC cells have been isolated from solid tumors in lungs, liver, head and neck, and pancreas as well as from melanoma and sarcoma [82]. However, CSC cells may be susceptible to infection with oncolytic viruses. For example, it was shown that the Zika virus of the Flaviviridae family infected glioblastoma stem cells [83]. Additionally, the adenovirus expressing the protein responsible for autophagy – Beclin-1 – was shown to induce autophagy in both CSCs and normal cancer cells [84].
A promising development in oncotherapy is the use of oncolytic viruses in combination with immunotherapy, which is carried out using monoclonal antibodies (mAb) that inhibit checkpoints serving as key regulators of the immune system. The first immunomodulator that showed a positive effect in the treatment of melanoma was the mAb ipilimumab – an inhibitor of cytotoxic T lymphocyte associated protein 4 (CTLA-4) that suppresses early activation and proliferation of T cells [85]. Pembrolizumab/nivolumab has been shown to suppress T-cells by inhibiting programmed cell death protein 1 (PD1) [86, 87]. The combination of HSV1-derived T-VEC with ipilimumab or pembrolizumab allowed for changing the antitumor immunity and enhancing the antitumor effect of T-VEC in the treatment of patients with brain melanoma metastases when traditional chemotherapeutic agents were ineffective [88, 89].
Thus, several advantages of the treatment of malignant neoplasms with oncolytic viruses in comparison to the existing types of therapy can be emphasized:
Unlike chemotherapy and radiation therapy, oncolytic viruses can selectively destroy tumor cells, including CSC, without targeting healthy cells due to local administration, higher selective targeting, and possible genetic modifications.
It is possible to use attenuated viruses that can penetrate the blood-brain barrier for the targeted destruction of distant metastases.
Oncolytic viruses can be adapted to the type of tumor and targeted by genetic modification, which opens up ample opportunity to create targeted anticancer treatment against tumors of various types and localizations.
A directed change of the gene expression in cancer cells can be accomplished through the treatment of tumors with oncolytic viruses, which can eventually lead to tumor regression after the infection. For this purpose, genetic elements, or entire cellular genes, such as the pro-apoptotic p53 protein gene, the apoptin protein gene, cytokines encoding genes, and the tumor necrosis factor gene, or genes encoding other factors that affect proliferation and/or cause programmed death of cancer cells, are artificially integrated into the viral genome [92-97].
Sequential application of a set of oncolytic viruses of different types – the sequential virotherapy – achieves a polyvalent effect enabling a deeper damage and eradication of the tumor and its metastases, as well as affecting the chemotherapy-resistant stem cancer cells.
Families of cytolytic viruses with oncolytic potential
The most studied viruses with oncolytic activity are adenoviruses and herpesviruses. Adenoviruses are known to have oncolytic activity against a large group of malignant tumors of diverse origin and different localization. To date, adenoviruses make up the largest group of viruses with oncolytic potential. They have also demonstrated a positive therapeutic effect on the largest number of malignant neoplasms of different nosologies. The oncolytic viruses from different viral families, their target tumors, and conducted clinical trials are presented in Table 1.
| Family | Viral oncolytic | Tumor specificity | References | |
|---|---|---|---|---|
| Strain | Design | |||
| Adenoviridae | ONYX-015 | Type 2/5, deletion of 827 bp in E1B gene, disrupting E1B 55K protein expression | Recurrent squamous cell carcinoma of the head and neck (Ph2)1
Adenocarcinoma of the pancreas (Ph1) Metastatic solid tumors (Ph1) Breast cancer Colorectal adenocarcinoma Hepatocellular carcinoma Melanoma Prostate cancer (Ph2) | [98] [99] [100] [101] [102] [103] [104] [58] |
| Oncorine (H101) | E1B deletion, E3 partial deletion | Squamous cell carcinoma of the head and neck (Ph3) | [105] | |
| DNX-2401 | Deletion of 24 bp in E1A, insertion of RGD-4C motif into the fiber protein | Glioma (Ph1) Glioblastoma (Ph1) | [106] | |
| VCN-01 | Insertion of RGDK motif, human hyaluronidase PH 20 | Pancreatic ductal adenocarcinoma Melanoma | [107] | |
| ONCOS-102 | Deletion of 24 bp in E1A, insertion of RGD, GM-CSF, replacement of Ad5 knob domain by Ad3 knob domain | Solid tumors (Ph1) Mesothelioma Ovarian cancer | [108] [109] [110] | |
| ICOVIR-7, ICOVIR-5 | Deletion of 24 bp in E1A, insertion of RGD-4C, modification of the E2F promoter Deletion of 24 bp in E1A, insertion of RGB, modification of the E2F promoter | Solid tumors Melanoma (Ph1) | [111] [112] | |
| CG0070 | Modification of E2F promoter, insertion of GM-CSF | Bladder cancer (Ph1, Ph2) | [113] | |
| Herpesviridae | Talimogene laherparepvec (T-VEC) | ICP34.5, ICP47 deletion, GM-CSF insertion | Melanoma, including melanoma with a mutation in the BRAF gene (licensed by FDA, EMA) Neuroendocrine tumors Breast cancer (Ph1) Squamous cell carcinoma of the head and neck | [114, 115] [116] [117] [118] |
| HF-10 | Lack of UL43, UL49.5, UL55, UL56, and LAT expression, overexpression of UL53, UL54 | Squamous cell carcinoma of the head and neck (Ph1, Ph2) Pancreas cancer (Ph1) | [119] [120] [183] | |
| G207 | ICP34.5 deletion, replacement of ICP6 by LacZ | Glioma (Ph1) | [121] | |
| SEPREHVIR (HSV1716) | ICP34.5 deletion | Solid tumors (Ph1) Neuroblastoma | [122] [123] | |
| M032 | IL12 expression | Glioma (Ph1) | [156] | |
| G47Δ | ICP34.5, ICP47 deletion, replacement of ICP6 by LacZ | Prostate cancer | [124] | |
| Paramyxoviridae | MV-CEA | CEA insertion | Ovarian cancer (Ph1) Breast cancer Colorectal cancer Prostate cancer Hepatocellular carcinoma Mesothelioma Myeloma Glioma | [125] [126] [126] [127] [128] [129] [130] [131] |
| MV-NIS | NIS insertion | Glioma | [130] [131] | |
| NDV V4UPM, MTH-68/H | NDV thermostable vaccine strain for poultry | Glioma | [132] [160] | |
| Picornaviridae | PVS-RIPO | Live attenuated type 1 (Sabin) poliovirus vaccine strain, insertion of internal ribosomal entry site (IRES) of HRV2 | Glioma | [133] |
| SVV-001 | Seneca Valley Virus, isolate 001 | Glioma Neuroendocrine tumors (retinoblastoma) Small cell lung carcinoma | [134] [135] | |
| Poxviridae | Pexastimogene devacirepvec (Pexa Vec/JX-594) | J2R gene replaced by hGM-CSF and lacZ | Colorectal cancer (Ph1) Hepatocellular adenocarcinoma (Ph1) Neuroblastoma (Ph1) Solid tumors (Ph1) | [136] [137] [138] [139] |
| GL-ONC1 | Insertion of three cassettes (Renilla luciferase–Aequorea green fluorescent protein, β-galactosidase, and β-glucuronidase), deletion of A56R, F14.5 L, and J2R genes. | Squamous cell carcinoma of the head and neck (Ph1) Peritoneal carcinomatosis (Ph1) | [140] [141] | |
| Reoviridae | Pelareorep (Reolysin) | Natural reovirus | Pancreatic ductal adenocarcinoma (Ph2) Breast cancer (Ph2) Colorectal adenocarcinoma Squamous non-small cell lung cancer Melanoma (Ph2) | [142] [143] [144] [145] [146] |
| Parvoviridae | H-1 PV | H-1 protoparvovirus (H-1PV) | Pancreatic ductal adenocarcinoma with peritoneal carcinomatosis | [147] |
| Rhabdoviridae | MG1MA3 | MAGE-A3 insertion | Glioblastoma (Ph2) | [148] [161] |
| VSV-IFNβ | Insertion of IFNβ, NIS genes | Myeloma Acute myeloid leukemia Squamous non-small cell lung cancer Endometrial cancer | [148] [148] [149] [150] | |
The largest number of developed oncolytic viruses are based on Ad5. The viral oncolytics ONYX-015, Oncorine (H101), and DNX2401 are in different phases (Ph) of clinical trials involving patients with head and neck squamous cell carcinoma, glioma, and pancreatic adenocarcinoma. The successful results of intratumoral administration of Oncorine (H101) in a Ph3 clinical trial allowed this oncolytic to be licensed in China [105]. Its subsequent use in a clinical trial against malignant pleural effusion resulted in complete recovery in 38% of the patients [151]. The oncolytics ORCA-010 and GC0070 showed promising clinical results. The main disadvantage of these oncolytic viruses, which can reduce their effectiveness, is the high level of vector-specific neutralizing antibodies in patients. This issue could be resolved by using chimpanzee adenovirus vectors or viruses of other types, as well as by the antigen shielding.
The large size of the HSV1 genome (about 150x103 bp) opens considerable opportunities for genetic modification of the virus. Removal of the genes that are not essential for replication allows for the insertion of transgenes that provide oncolytic properties. The first oncolytic candidate – the T-VEC virus (Talimogene laherparepvec, commercial name Imlygic) – was officially approved by the FDA and EMA for the treatment of patients with inoperable melanoma. The T-VEC oncolytic was created on the basis of HSV1 virus with the deletion of the genes encoding the infected cell proteins (ICP) ICP34.5 and ICP47 and the insertion of two copies of the human protein hGM-CSF gene which increases the presentation of the tumor antigen by dendritic cells [152, 153]. As a result, cancer but not healthy cells are infected by the virus [114]. There are ongoing clinical trials (Ph1) on the use of T-VEC alone or in combination with ipilimumab/pembrolizumab for the treatment of patients with squamous cell carcinoma. Another viral oncolytic of the Herpesviridae family is the spontaneous HF-10 mutant, whose genome lacks the UL43, UL49.5, UL55, UL56, and LAT genes, which ensures active reproduction of the virus in cancer cells [119, 120, 154]. Due to the enhanced induction of CD4+, CD8+, and NK cells, HF-10 effectively suppresses tumor growth, as it was demonstrated against melanoma, pancreatic adenocarcinoma, squamous cell carcinoma of the head and neck, and breast cancer on intratumoral and peritoneal administration [155]. The second generation of oncolytic viruses derived from HSV1 includes the M032 virus expressing interleukin 12 (IL12), which is being tested (Ph1) for the treatment of glioma [156]. G47Δ is the third-generation oncolytic virus with a deleted ICP47, unlike the G207 virus [124]. Currently, research is underway to create genetic variants of the herpes virus, and a wide range of targeted viral vectors has already been created. Thus, more than 20 viral strains, which are currently at different stages of clinical research, have been developed only for the treatment of glioblastoma tumors [157]. New candidates, such as ONCR-177, contain the genes expressing IL12, CCL4, and FLT3LC proteins and include anti-CTLA-4 and PD1 blocking sequences to increase NK and T cells activation and DC availability as well as to prevent depletion of T cells. Candidate C134, which contains the human cytomegalovirus IRS1 gene in addition to the ICP34.5, was created to increase the activity of the oncolytic virus against glioma [158]. The genome of another virus, RP1, was modified by adding the genes that encode GM-CSF, and gibbon ape leukemia virus fusogenic membrane protein GALV-GP/R. Further modification (virus RP2) was made to express anti-CT-LA-4 to enhance antitumor activity against squamous cell carcinoma [159].
The vaccine strains of paramyxoviruses, picornaviruses, poxviruses, and adenoviruses are promising for the development of new oncolytic viruses. The viruses of these families (both vaccine strains and their wild-type precursors) are capable of replicating in malignant cells of neurogenic origins, which makes them promising candidates for the development of new oncolytic viruses for the treatment of brain tumors and cerebral metastases [160, 161]. It was shown that various genetically modified viruses based on the vaccine strains are active against a broad range of malignant neoplasms (Table 1). A viral oncolytic from the Paramyxoviridae family (MV-CEA) has passed the clinical trials (Ph1) involving patients with ovarian cancer.
An oncolytic from the Poxviridae family JX-594 (known as Pexa Vec) has successfully passed the clinical trials (Ph1) involving patients with refractory metastatic colorectal cancer, hepatocellular adenocarcinoma, neuroblastoma, and solid tumors. However, the clinical trials (Ph3) in patients with advanced hepatocellular carcinoma showed no clinical efficacy of this oncolytic and were discontinued. The clinical trials (Ph1) of the viral oncolytic of the Poxviridae family GL-ONC1 in patients with head and neck squamous cell carcinoma and peritoneal carcinomatosis are ongoing.
Reoviruses infect tumor cells with the activated prooncogenic Ras-dependent signaling pathway, which provides selective inhibition of protein kinase R (PKR) phosphorylation, thus promoting virus reproduction [162]. The oncolytic Reolysin® (later named Pelareorep), which showed activity against many malignant tumors, was developed based on the reovirus type 3. An important property of the reovirus is its ability to cross the blood-brain barrier and penetrate cells of both the primary tumor and metastases in the brain [163, 164]. Reolysin® is in clinical trials (Ph2) in patients with pancreatic ductal adenocarcinoma, breast cancer, and melanoma.
Influenza A viruses – vaccine strains or avian influenza viruses – are good candidates for the treatment of malignant tumors. In a pilot experiment, a positive therapeutic effect was shown for the virus with an altered gene encoding the NS1 protein in a model of intradermal melanoma implants in mice, as well as against non-small-cell lung cancer in experiments in transgenic immunocompetent mice [41, 165]. By the 12th day after infection, the amount of tumor mass in the lungs of mice decreased by 70% compared with the uninfected animals [165]. The defect in the viral NS1 protein led to disruption of the cellular IFN response and promoted the activation of tumor cell apoptosis, which implied the increased proinflammatory and antitumor effect of the virus with the modified NS1 gene [27, 39].
VSV of the Rhabdoviridae family was shown to infect cells with a defective IFN signaling pathway, including cancer cells, subsequently causing their lysis by apoptosis. The oncolytic viruses based on VSV were shown to be active against non-small-cell lung cancer, endometrial cancer, and leukemia (T-cell leukemia, acute myeloid leukemia) in experimental models [148-150]. The tumor specificity of viruses from the rhabdovirus and parvovirus families is shown in Table 1.
Clinical trials of oncolytic viruses against various nosological forms of cancer
Most clinical studies of oncolytic viruses have been carried out against melanoma and malignant neoplasms of the gastrointestinal tract. As a rule, clinical trials are conducted in groups of patients with stage III and IV cancer as well as in patients with metastases and relapses of the disease. Melanoma (stages III and IV), glioma, and adenocarcinoma of the pancreas have a poor prognosis and lead to a high mortality rate in patients. Glioma and pancreatic adenocarcinoma are often diagnosed in the late stages of the disease when the standard methods of therapy are ineffective. Maximal progress in the use of viral oncolytics has been achieved against these nosological forms of cancer. This may explain the large number of conducted clinical trials [166].
Melanoma treatment
Melanoma (melanocarcinoma, melanosarcoma) is a malignant tumor that develops from melanin-producing cells – melanocytes and melanoblasts. These tumors are localized mainly on the skin, less often on the retina and mucous membranes; they are often recurrent and quickly metastasize to distant organs through the lymphogenous and hematogenous pathways. Melanoma is characterized by a wide variety of histological structures and clinical manifestations. Surgical excision of melanoma can cure the tumor in the early stages of the disease but is ineffective for the treatment of metastatic melanoma. Melanoma responds poorly to radiation and chemotherapy. The dacarbazine treatment regimens used in clinical practice give an objective response rate (ORR – the proportion of the patients who have an objective response (total response: complete + partial + stabilization)) only in 15% of cases and prolong remission by five years. For example, in metastatic melanoma without mutations in the BRAF gene, immune checkpoint inhibitors (ipilimumab, pembrolizumab, and nivolumab) are used as the first-line therapy. The treatment with nivolumab and ipilimumab results in an ORR of 18% [167]. The estimated five-year survival rate for patients with early melanoma is 98%, after the spread of melanoma to the lymph nodes – 68%, and after metastasis to distant organs – 30%. The melanoma mortality rate is 15-20%. According to the American Cancer Society, the life expectancy of patients with stage III and IV metastatic melanoma is about four months without treatment, and 6-8 months after standard treatment (surgery, chemotherapy) [168].
The oncolytic Imlygic® (T-VEC) successfully passed three phases of clinical trials in patients with melanoma and was approved by the FDA and EMA for clinical use in 2015 [16]. Clinical trials were performed in a group of patients with stage III and IV metastatic melanoma (according to the Classification of Malignant Tumors, TNM and American Joint Committee on Cancer, AJCC; Table 2). The virus was injected into the tumor or subcutaneously. Nine HSV seronegative patients with metastatic melanoma were involved in the Ph1 trial. A stable positive effect was observed in two cases [169]. The clinical trial (Ph2) was performed in a group of 50 patients with inoperable melanoma (stages IIIc-IV). The observed ORR was 26% including eight patients with a complete response and five with a partial response. The overall survival rate (OSR), defined as the percentage of patients alive during a selected period after diagnosis, reached 58% in one year and 52% in two years [170]. The clinical trial (Ph3) was conducted in a group of 436 patients with melanoma (IIIb-IV). The durable response rate (DRR) reached 16.3%. The median overall survival (mOS) – the length of time from the start of treatment or from the moment of diagnosis, that half of the group of the diagnosed patients remain alive – was 23.3 months [115]. Additionally, treatment with T-VEC was studied in combination with anti-CTLA-4 (ipilimumab) and anti-PD1 (pembrolizumab) monoclonal antibodies in patients with a progressive stage IIIb and IV melanoma. In clinical trial (Ph1) in a group of eight patients using the combination of T-VEC therapy with ipilimumab, the ORR reached 50% and the OSR – 67% in 1.5 years [170]. The Ph2 trial included 198 patients and resulted in the ORR of 39% (the ORR of ipilimumab monotherapy is only 18%) [167]. The combination therapy in 21 patients using oncolytic T-VEC with pembrolizumab (Ph1) showed the ORR of 48% [171]. Thus, Imlygic® proved to be an effective oncolytic for the treatment of advanced melanoma, while its use in combination with immunotherapy increased the total treatment efficiency.
| Oncolytic virus | Clinical trial data | ||||
|---|---|---|---|---|---|
| Phase of clinical trials, Ph | Number of patients | Tumor type, stage | Administration route | ORR (%), mOS (months) | |
| T-VEC | Ph1 | 9 | Metastatic melanoma | i.t. | 2 patients with a stable positive effect |
| Ph2 | 50 | Melanoma IIIc, IV | i.t. | 26%, 12 months | |
| Ph3 | 436 | Melanoma IIIb, IIIc, IV | 295 patients, i.t. 141 patients, s.c. | 26.4%, 23.3 months | |
| HF10 (Canerpaturev-C-REV) | Ph1 | 28 | Metastatic melanoma | i.t. | 66.7%, 34.6 months |
| Ph2 (+ipilimumab) | 46 | Metastatic melanoma | i.t. | 41%, 21.8 months | |
| Coxsackievirus A21 (CAVATAK) | Ph2 | 57 | Melanoma IIIc, IV | i.t. | 28%, 6 months |
| Pelareorep (Reolysin) | Ph2 Ph2 (+carboplatin/ paclitaxel) | 21 14 | Metastatic melanoma | i.v. | 21%, 5.2 /10.9 months |
| Vaccinia GM-CSF (Pexa-VEC) | Ph1 | 12 | Melanoma IV | i.t. | 21% |
i.t. – intratumoral injection, i.v. – intravenous injection, s.c. – subcutaneous injection
Two phases of the clinical trials with the attenuated viral oncolytic HF-10 were completed in patients with melanoma. In a Ph2 study, 46 patients with stage IIIb-IV melanoma were treated with HF-10 and ipilimumab combination. The ORR reached 41%, and the mOS was 21.8 months (the ORR with ipilimumab monotherapy is only 18%) [172, 173]. Thus, HF-10 is a promising viral oncolytic for the treatment of melanoma, especially in combination with immunotherapy.
Two phases of the clinical trials with a reovirus-based oncolytic Pelareorep (Reolysin) were completed in patients with melanoma using intravenous administration. In the Ph2 clinical trial involving 21 patients, Reolysin monotherapy proved to be ineffective, since the oncolytic induced the production of neutralizing anti-reo-virus antibodies (NARA) that may have limited the efficiency of viral replication and the oncolytic effect [174, 175]. The clinical study Ph2 (14 patients with advanced melanoma) with Reolysin/Carboplatin (Paclitaxel) combination therapy showed the ORR of 21%, while the mOS reached 5.2 months (Reolysin+Carboplatin) and 10.9 months (Reolysin+Paclitaxel). With Carboplatin or Paclitaxel monotherapy in the other clinical trials, the mOS reached three and nine months respectively [176, 177]. Therefore, as it follows from the presented data, the use of reoviruses for the treatment of melanoma requires additional investigations.
The clinical trials with a recombinant Vaccinia-GM-CSF virus have been conducted in patients with metastatic melanoma. The virus replicated well in cancer cells, causing inflammatory lymphocytic infiltration and tumor necrosis [178]. In the Ph1 trial (12 patients), the intratumoral administration of the oncolytic resulted in the 21% ORR and led to an increase in the T cell immune response to the melanoma-specific antigens (gp100 and MART-1) [178]. More studies are needed for the development of efficient VV-based oncolytics for the treatment of advanced melanoma.
Glioma treatment
Glioma is a brain tumor. The main treatment for gliomas is surgery combined with radiation and chemotherapy. There are a few clinical trials involving patients with gliomas due to the development of complications after surgical operations (changes in mental status, speech impairment, cerebral edema). The 10-year survival rate for a low-grade glioma is 47% and the mOS is 11.6 years [179]. Due to the aggressive course of the disease, the mOS of patients with a grade III glioma is three years, and with a grade IV glioma – from 6 to 15 months [180].
The HSV-derived G207 oncolytic passed a Ph1 clinical trial (27 patients with glioblastoma), showing the mOS of 6.6 months (Table 3). With a combination of radiotherapy and G207 treatment, the mOS reached 7.5 months [181]. However, this effect is comparable to the results of the standard treatment regimens for glioma without the use of oncolytic viruses.
| Oncolytic virus | Clinical trial data | ||||
|---|---|---|---|---|---|
| Phase of clinical trials, Ph | Number of patients | Tumor type, stage | Administration route | mOS (months) | |
| HSV 1716 (Seprehvir) | Ph1 | 9 | Malignant glioma | i.t. | 24 months |
| Ph2 | No data | Recurrent glioma in children | i.t. | No data | |
| G207 | Ph1a | 21 | Glioblastoma | i.t. | 6.6 months |
| Ph1b | 6 | Glioblastoma | i.t. | 6.6 months | |
| ONYX015 | Ph1 | 24 | Malignant glioma | i.t. | 6.2 months |
| DNX-2401 | Ph1 | 25 | Recurrent malignant glioma | i.t. | 5 patients 36 months |
| Reolysin | Ph1 | 12 | Recurrent malignant glioma, III, IV stages | i.t. | 1 patient 5 months 1 patient 54months |
| NDV | Ph1 | 11 | Recurrent glioblastoma | i.v. | 7 months |
i.t. – intratumoral injection, i.v. – intravenous injection
In a Ph1 trial (25 patients with recurrent malignant glioma), the Ad-based oncolytic DNX-2401 demonstrated the mOS of 36 months. At the same time, the tumor size decreased by 95% in three patients [106]. That exceeds the results of the standard treatment, although additional studies are required for a wider DNX-2401 application. Additionally, an oncolytic based on NDV (strain HUJ) showed the mOS of 7 months in a Ph1 study in 11 patients with recurrent glioblastoma, which corresponds to the results of the standard treatment [132].
Thus, the treatment of gliomas with viral oncolytics showed promising results and requires further confirmation in Ph2 and Ph3 studies.
Treatment of ductal adenocarcinoma of the pancreas
Ductal adenocarcinoma of the pancreas is a malignant neoplasm that is difficult to treat. The mortality from this type of cancer in Russia is 3.4% [2]. The main methods of treatment are surgical resection, radiation, and chemotherapy. The prognosis is poor since the disease is usually diagnosed at a late stage. Immunosuppressive factors such as Treg, M2 (tumor-associated macrophages), IL10, and transforming growth factor β (TGFβ), accumulate in the tumor microenvironment, thus limiting the effectiveness of chemotherapy and immunotherapy for pancreatic cancer. The five-year survival rate of patients with this disease is 6% [1]. The clinical trials of ipilimumab and gemcitabine in patients with pancreatic ductal adenocarcinoma showed the ORR of 10.8% with the mOS of 10 months [182].
Oncolytic viruses (herpesviruses and reoviruses) have shown an antitumor effect against pancreatic cancer (Table 4) [120, 183]. According to the Ph2 trial in 34 patients with pancreatic ductal adenocarcinoma, the treatment with the viral oncolytic Reolysin leads to the mOS of 10.2 months [142]. These results do not differ from the results of monoclonal antibody therapy. Reolysin may be considered a promising candidate for cancer treatment, although more studies are needed including clinical trials with a larger number of patients. Currently, there are ongoing Ph1 clinical trials of the oncolytic viruses ONYX015, T-VEC, and H-10 in patients with pancreatic adenocarcinoma.
| Oncolytic virus | Clinical trials data | ||||
|---|---|---|---|---|---|
| Phase of clinical trials, Ph | Number of patients | Tumor type, stage | Administration route | mOS (months) | |
| ONYX015 | Ph1 | 23 | Inoperable adenocarcinoma | CT controlled injection | No data |
| Ph2 (+gemcitabine therapy) | 21 | Progressive and metastatic adenocarcinoma | i.v., endoscopic probe, gemcitabine therapy | 6 patients, partial tumor regression | |
| H10 | Ph1 | 8 | Pancreatic adenocarcinoma | i.t., through the catheter in the course of surgery | 3 patients, partial tumor regression, 6 months |
| T-VEC | Ph1 | 17 | Pancreatic adenocarcinoma | No data | No data |
| Reolysin | Ph2 | 34 | Pancreatic adenocarcinoma | i.v. | 10.2 months |
i.t. – intratumoral injection, i.v. – intravenous injection
Thus, a number of viral oncolytics based on the herpes virus or adenovirus have successfully passed clinical trials and showed a positive therapeutic effect against inoperable melanoma and squamous cell carcinoma of the head and neck in the absence of pronounced side effects and general toxicity. One oncolytic virus can have a positive therapeutic effect on several histological forms of a tumor, and a stronger therapeutic effect can be achieved by combining viral oncolytics with immunotherapy.