
INTRODUCTION
Neurodevelopmental disorders (NDDs) are classified as heterogeneous disorders having diverse phenotypic presentations that arise during the developmental period and are mainly characterized by cognition impairment and defective behavioral and psychomotor skill development. These disorders are caused by changes in neural mechanisms and brain structure due to defective development. The prevalence of these disorders has been estimated to be >3% worldwide ( Gilissen et al., 2014). NDDs have a diverse etiology as they constitute intellectual disability (ID), communication disorders, autism spectrum disorder (ASD), attention deficit/hyperactivity disorder (ADHD), and several other neurodevelopmental motor disorders ( Elsabbagh et al., 2012). Microcephaly has a heterogeneous etiology with environmental and genetic factors ( Kaindl et al., 2010; Nicholas et al., 2010). The prevalence of epilepsy is high in microcephaly patients. Due to the clinical diversity and genetic complexity of NDDs, the diagnosis is time-consuming and expensive.
Several Genetic and environmental factors have been reported to be the etiological factors of NDDs ( Ehninger et al., 2008). Still, the primary causal factor for these disorders is genetic aberrations, the recognition of which is key for precise (early) diagnosis as well as proper genetic counseling of patients and their caregivers for appropriate patient management ( Niemi et al., 2018). These genetic aberrations mainly occur due to consanguineous marriages (for autosomal recessive phenotypes) or de novo autosomal dominant variants in the offspring of unaffected parents. Several NDD-causing genes have been classified as highly susceptible genes, and their mutations pose a significant risk of developing NDDs ( Iossifov et al., 2015; Hautakangas et al., 2022). Similarly, fewer genes run in families, and their mutations are associated with developing NDDs. The epistemic interactions and the mutational load of these less vulnerable genes can lead to the development of a complex NDD ( Parenti et al., 2020). To date, >1500 genes have been reported to be responsible for NDDs ( Leblond et al., 2021); however, more research is still required due to the polygenic nature of these disorders. Furthermore, the risk of developing NDDs has been found in genes that are protein synthesis regulators, transcriptional regulators, epigenetic regulators, and those that are associated with synaptic signaling ( Zug, 2022). Several large-scale genomic studies have facilitated the identification of de novo mutations in the protein-coding genes ( Baasch et al., 2014; Cardoso et al., 2019).
Primary microcephaly (MCPH, OMIM 251200) is a congenital genetic NDD characterized by a small brain size along with mild to moderate ID. The head circumference (HC) is reduced from −4 standard deviation (SD) to −2 SD at the time of birth below the age and sex means as compared with the normal HC Patient with microcephaly came up with different level of ID ( Woods et al., 2005; Kaindl et al., 2010). MCPH symptoms include seizures and delays in the development of gross motor, cognitive, language, and visual motor skills ( Nawathe et al., 2018).
The incidence of MCPH varies with population, ranging from 1:30,000 to 1:250,000 per live birth ( Zaqout et al., 2017). MCPH is more prevalent among Asian and Middle Eastern populations than among white people ( Muhammad et al., 2009). So far, 18 loci and residing genes have been reported to have variations in the genes for MCPH ( Faheem et al., 2015).
Biallelic alterations in ASPM are the most frequent etiology of MCPH (68.6%), followed by changes in WDR62 quality (14.1%) and MCPH1 gene (8%). Given the lack of mutations in the known loci in roughly 50-75% of Western Europeans or North Americans with MCPH and roughly 20-30% of Indians or Pakistanis with MCPH, further hereditary loci are still expected to exist ( Hussain et al., 2013; Naveed et al., 2018). Primarily due to the high proportion of consanguineous marriages, genetic diseases with recessive modes of transmission are prevalent in some isolated groups living in rural Pakistan. Due to the relatively high birth rate in certain parts of Pakistan, families tend to be larger and include more afflicted people. This study offers a great chance to map and pinpoint the mutations that cause MCPH. We may infer from linkage analysis that the ASPM gene is a significant genetic factor for autosomal recessive MCPH in Pakistan. More research is required for additional molecular clarifications to lower the risk of the disease. Thus, prenatal diagnosis and mutation screening in Pakistan’s MCPH families can help lower the prevalence of MCPH in the consanguineous population. The factors that account for the origin of microcephaly in humans are yet unknown.
In the current study, we describe two families exhibiting features of microcephaly and NDDs. In addition, whole-exome sequencing (WES) revealed previously reported variants in ASPM gene that confirm the molecular diagnosis in two families.
METHODS
Research subjects and study approval
All the families having NDDs were evaluated using standard methods in the hospitals. The study was accomplished based on standard Helsinki protocols, and the institutional ethical review board approved the study. Signed informed consent for the research study and publication of the research data was obtained from the parents. Clinical evaluations were performed in local government hospitals.
Genomic DNA isolation
Peripheral blood was obtained in Ethylenediamine tetraacetic acid (EDTA) vacutainers from all the available individuals in each family and presented with asterisks in the pedigrees ( Fig. 1a and b). DNA was extracted and quantified using standard methods ( Irfanullah et al., 2015; Umair et al., 2016; Younus et al., 2019).
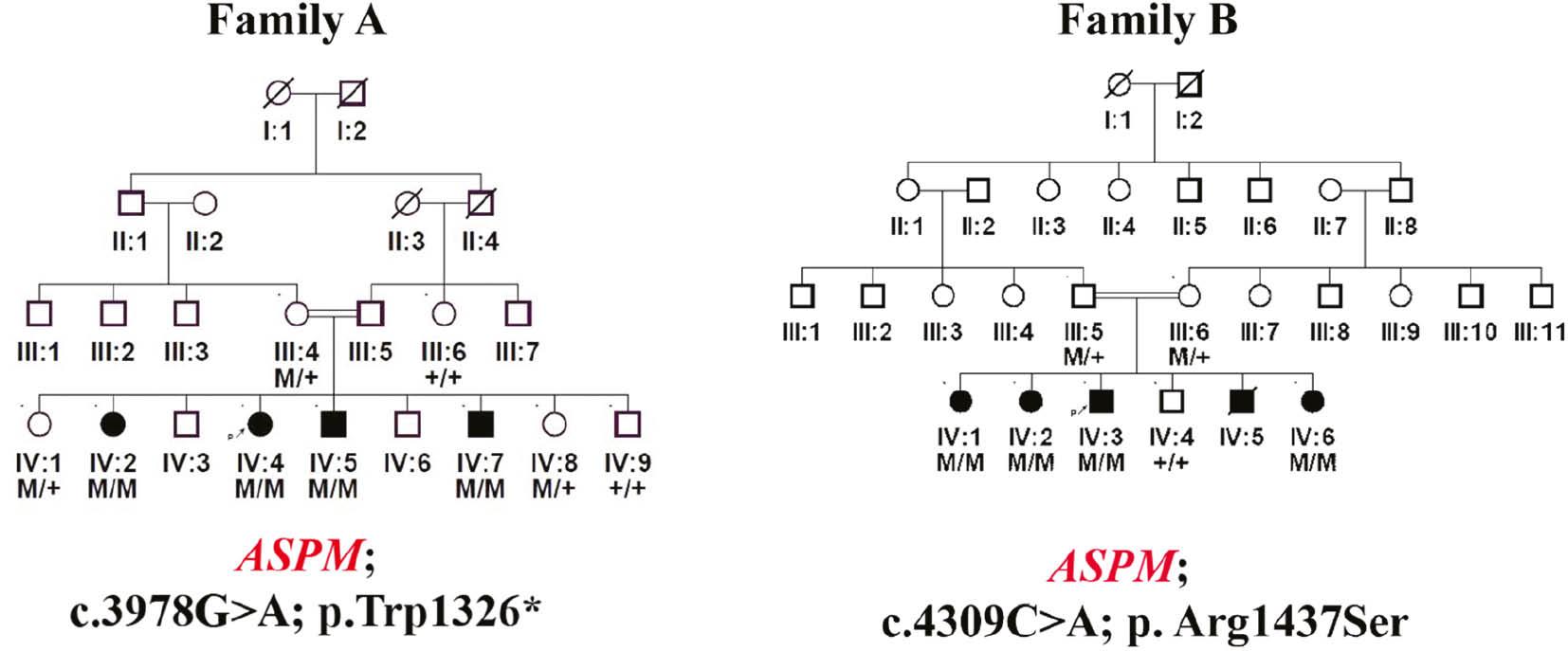
(a) Four generation pedigree of family A showing a homozygous nonsense variant (c.3978G>A; Trp1326*) in exon 17 in the ASPM gene in all the affected individuals of the family. (b) Four generation pedigree of family B showing a missense variant (c.4309C>A; p. Arg1437Ser) in exon 18 of the ASPM gene in all the affected individuals of the family. Double lines indicate consanguineous union; filled circles and squares represent affected individuals; crossed circle and squares indicate deceased individuals.
WES and data analysis
DNA of a single affected individual from each family was subjected to WES using Illumina HiSeq4000 with 150 bp paired-end reads yielding an average of ×60 coverage per targeted base using a ligation-mediated di-base detection system as previously described ( Umair et al., 2017, 2018, 2019). All the reads obtained were aligned to human assembly hg19 (GRCh37) using Burrows–Wheeler Aligner (v 0.7.5), and PINDEL, SAM tools, and Exome Depth were used for variant calling. Subsequently, the final variant calling format file was generated that was uploaded and analyzed using the Illumina basespace online tool ( Ullah et al., 2019; Umair et al., 2020). Pathogenic, likely pathogenic, and variants of uncertain significance (VUSs) were searched according to the college of medical genetics and genomics (ACMG) guidelines.
Variant classifications and prioritization
Variants were classified according to the ACMG guidelines. Variant filtration was performed using standard methods ( Umair et al., 2020; Nawaz et al., 2021a, b). Different online databases (ExAC, gnomAD, 1000 genomes, etc.) were used to exclude the polymorphic nature of the identified variants. In addition, the variants were fully segregated among available family members. Conservation of the mutated amino acids was observed using national CENTRE for bio-informatics (NCBI) homologene ( http://www.ncbi.nlm.gov/homologene/) ( Fig. 2a and b).

Pathogenicity of the identified variants
The pathogenicity index for the detected sequence variant was calculated using Mutation Taster, Polymorphism Phenotyping V2 (PolyPhen-2), and Sorting Intolerant from Tolerant. The frequency of the variant in the general population was determined using ExAC, gnomAD, 1000 genomes, and in-house control exomes.
Sanger sequencing
The variant obtained after WES data analysis was Sanger sequenced in all the available members of the family as described previously ( Hayat et al., 2020; Nawaz et al., 2021a, b; Khan et al., 2023). Primers were designed using Primer 3 and/or Exon Primer for all the identified variants and will be provided upon request.
RESULTS
We enrolled two consanguineous families having seven affected individuals exhibiting hallmark features of microcephaly and additional NDD phenotypes with a likely autosomal recessive inheritance pattern. The affected individuals were clinically and genetically evaluated. The local hospitals performed detailed clinical examinations, and molecular analysis was performed in standard laboratories. Affected individuals from each family showed various types of phenotypical representation.
Family A
The affected individuals in family A include two girls and two boys, aged between 8 and 15 years. They revealed hallmark features of primary microcephaly and developmental delay. The brain magnetic resonance imaging (MRI) of IV-4 revealed prominent spaces in the cerebrospinal fluid (CSF). The differential includes benign enlargement of the extra-axial CSF space versus brain parenchymal volume. The appearance definitely shows genuine brain parenchymal volume loss ( Fig. 3a).
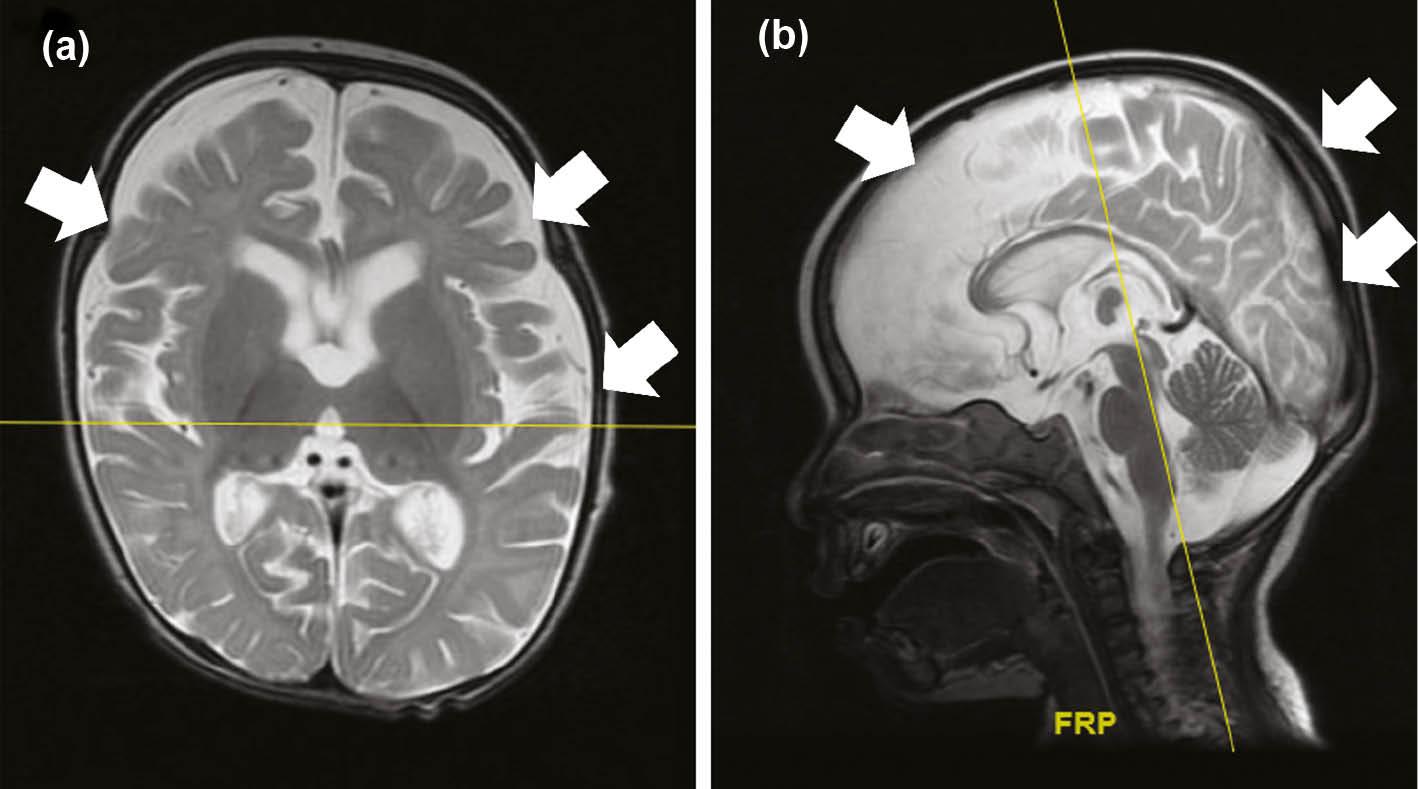
(a,b) Brain MRI of the affected individuals in family A and family B. (a) Brain MRI of the affected individual in family A revealed parenchymal volume loss in different parts of the brain suggesting severe changes that might lead to ID. (b) Brain MRI of the affected individual (IV-3) in family B revealed delay in white matter myelination and underdeveloped corpus callosum that can be associated with the severity of the disorder. Abbreviation: ID, intellectual disability.
Family B
The affected individuals in family B were three girls and two boys, aged between 12 and 22 years. All the affected individuals (IV-1, IV-2, IV-3, IV-5, IV-6) revealed microcephaly and NDD. The HC of all the affected individuals was less than normal. The MRI of IV-3 revealed delay in white matter myelination and underdeveloped corpus callosum ( Fig. 3b).
WES and filtration steps
DNA of a single affected individual from each family was subjected to WES using an Illumina HiSeq 4000 system as previously described ( Umair et al., 2021). After WES, the variants obtained were filtered systematically using standard methods to screen the data and identify the disease-causing variant in a gene. The genes and variants were cross-matched with OMIM, ClinVar, and human gene mutation database (HGMD) (HGMD® Professional 2022.2) for the disease gene–association, and either the variant is already reported in the literature or novel ( Fig. 4). During the filtration steps, we first focused on pathogenic variants (Class 1) and likely pathogenic variants (Class 2). After that, if the variant was not rare and associated with the disease phenotype, we moved toward VUSs (Class 3).
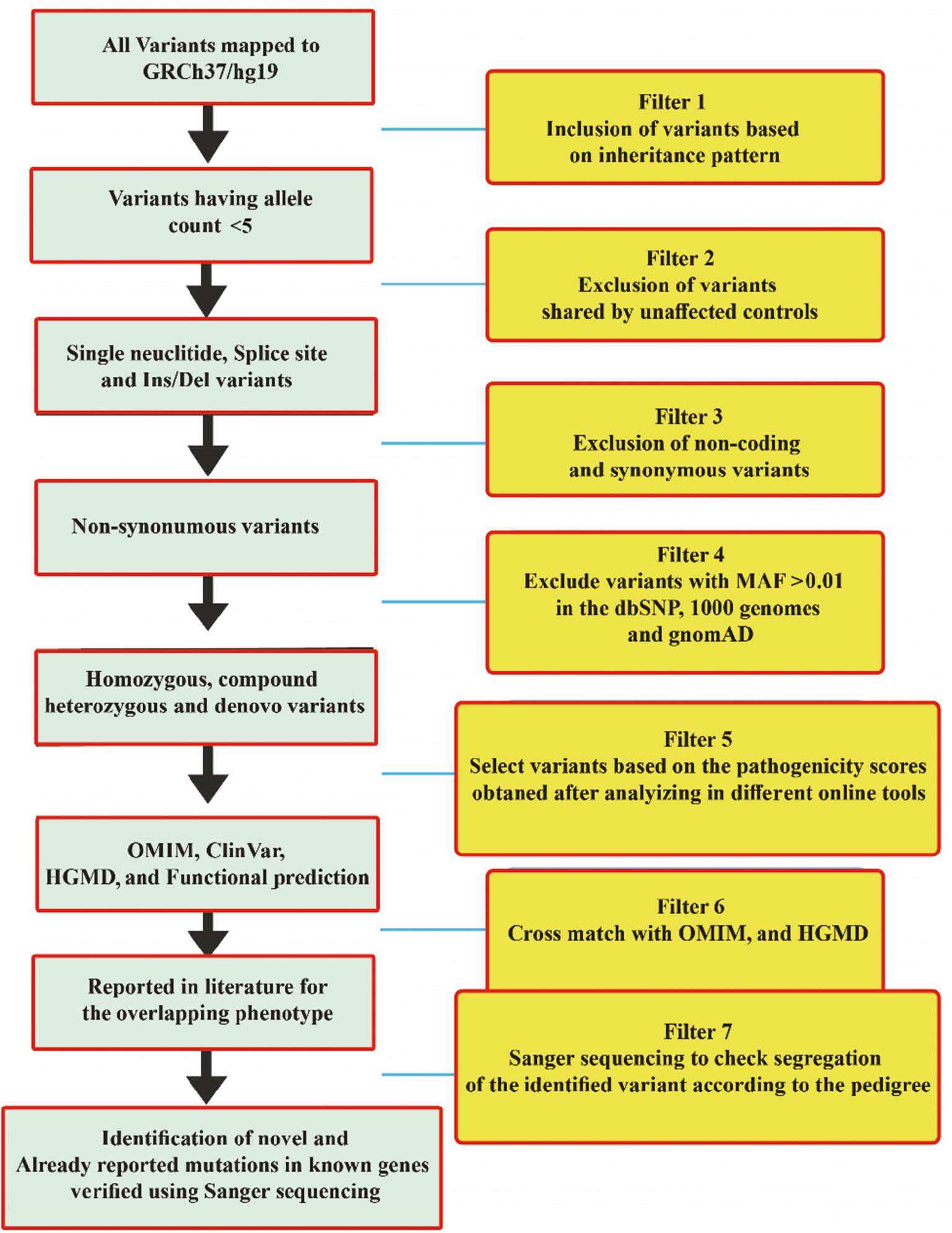
Different steps used for the filtration of variants in whole-exome data. Starting with the inheritance pattern of the diseases (pedigree) and ending with the Sanger sequencing of the variants in all family members.
In the present study, two families were evaluated. In family A, we identified an already reported homozygous nonsense variant (c.3978G>A; Trp1326*) in exon 17 of the ASPM gene (NM_018136.5), that was classified as pathogenic (Class1) according to the ACMG classification criteria. The identified variant [c.3978G>A] showed perfect segregation in all the available family members and the mutated Trp amino acid was highly conserved across different species.
While in family B, we identified an already reported missense variant (c.4309C>A; p. Arg1437Ser) in exon 18 of the ASPM gene (NM_018136.5), that was also classified as a VUS (Class 3) according to the ACMG classification criteria. The identified variant showed perfect segregation in all the available family members and the mutated Trp amino acid was highly conserved across different species. ASPM is located on chromosome 1q31.3 and consists of a total of 28 exons, encoding a 3478 amino acid protein.
Mutations validation
The identified mutation was not observed in gnomAD, ExAC, 1000 Genomes, and in-house exomes. The sequence variations/mutations detected in the present study were characterized using the Human Genome Variation Society ( http://www.hgvs.org/mutnomen/) nomenclature. Identified variants were classified as already reported variants using online available databases ( Sharo et al., 2023).
DISCUSSION
NDDs are a diverse group of diseases associated with different clinical characteristics, such as severity, onset age, prognosis, and reactions to treatment. NDDs are a heterogeneous group of disorders associated with ID, global developmental delay, epilepsy, mild-to-severe microcephaly, ASDs, ADHD, and learning disorders lacking precise boundaries in their clinical definitions, epidemiology, genetics, and other associated phenotypes that might result in significant limitations in intellectual functioning and adaptive behavior ( Morris-Rosendahl and Crocq, 2020; Parenti et al., 2020). Additional features such as hearing impairment, speech and language disorders, ID, epilepsy, and learning disorders were commonly observed in NDD patients ( Francés et al., 2022). NDD prevalence is estimated at 1-3% of the world population; however, different countries and ethnicities have different prevalence depending on the culture, social interactions, and consanguineous marriages. The prevalence of NDDs in children aged 2 to 6 years ranged from 2.9% to 18.7%, while children aged 6 to 9 years showed 6.5% to 18.5% prevalence. Additional features such as hearing impairment, speech and language disorders, ID, epilepsy, and learning disorders were commonly observed in NDD patients ( Francés et al., 2022). However, genomic variations play a major role in the susceptibility of diseases, the age of disease onset, and treatment options for various classes of neurological diseases ( Foo et al., 2013). Similarly, almost 40% of the ID conditions remain equivocal and almost ˜50% have an environmental etiology such as improper care during pregnancies, multiple pregnancies with less gap, poor nutrition, brain ischemia (prenatal/perinatal), poor medical services, and postnatal infections. The ID having a genetic etiology, such as mutations in genes and chromosomal abnormalities, is rare yet having a huge impact on the family and economy.
In the present study on families A and B, the WES followed by Sanger sequencing revealed a G to A transition at nucleotide position 3978, producing an immediate premature stop codon (Trp1326*) in exon 17 of the ASPM gene in family A, while in family B c.4309C>A; p.Arg1437Ser in the ASPM gene. Both the variants were further confirmed using bi-directional Sanger sequencing.
This nonsense variant might result in NMD and production of a truncated nonfunctional ASPM protein. The mutation (p.Trp1326*) is the hot-spot sequence variant in the Pakistani population ( Kousar et al., 2010). According to Saadi et al. (2009), augmented reality (AR) MCPH are mostly caused by bi allelic ASPM mutations. ASPM plays a significant role in the orientation of the mitotic spindle between symmetric and asymmetric divisions, hence regulating brain size ( Fish et al., 2006; Higgins et al., 2010). In people with MCPH with an ASPM gene mutation, this modulation of mitotic spindle orientation may be a key mechanism regulating brain size ( Bienz, 2001). According to a study of their phenotypes, the two ASPM gene variations known to cause microcephaly and NDDs have phenotypic overlap with previously described entities. The impacts of genetic modifier variations or the varying effects of allelic mutations, which may be particularly significant in populations with a high consanguinity rate, can cause some aberrations to develop.
In the recent past, investigation of primary microcephaly and NDDs from a genetic, psychological, neuroanatomical, and molecular perspective discovered unique genes and identified related pathways. Molecular genetics and developmental cognitive neuroscience are now being integrated, which is a result of enormous breakthroughs in all fields of study ( Deng et al., 2023; Meng et al., 2023; Umair, 2023). Our understanding of developmental brain disorders will be improved by discovering population-wide variations and novel related genes ( Asiri et al., 2020; Khan et al., 2020; Nøstvik et al., 2021; Umair et al., 2021). Additionally, in order to prevent severe microcephaly and NDDs from impacting future generations, prenatal genetic screening and prenatal screening are necessary ( Alfadhel et al., 2019; Alyafee et al., 2021a, b; Alyafee et al., 2022). Rare genetic disorders such as NDDs should be prevented before they become common.
In the future, rare genetic and multifactorial disorders will likely be managed using a combination of cutting-edge technologies, customized medical strategies, and a deeper understanding of genetics, epigenetics, and molecular pathways. Gene therapy for rare genetic conditions shows great potential for treating a number of disorders, but its acceptance will rely on a number of conditions, including technologies like CRISPR-Cas9-based gene editing, personalized medicine, enzyme/targeted therapy, and research collaborations ( Umair, 2023; Umair and Waqas, 2023).
Our study demonstrates the clinical utility of WES for NDDs in families having frequent consanguineous, to elucidate the molecular basis of very heterogeneous recessive disorders. Uncovering variants in a reported gene, correct molecular diagnosis, and identification of candidate genes for such conditions in inbred families will trigger the identification of matching mutations in other populations.