- Record: found
- Abstract: found
- Article: found
On the Importance of Polar Interactions for Complexes Containing Intrinsically Disordered Proteins
Read this article at
Abstract
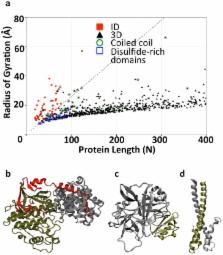
There is a growing recognition for the importance of proteins with large intrinsically disordered (ID) segments in cell signaling and regulation. ID segments in these proteins often harbor regions that mediate molecular recognition. Coupled folding and binding of the recognition regions has been proposed to confer high specificity to interactions involving ID segments. However, researchers recently questioned the origin of the interaction specificity of ID proteins because of the overrepresentation of hydrophobic residues in their interaction interfaces. Here, we focused on the role of polar and charged residues in interactions mediated by ID segments. Making use of the extended nature of most ID segments when in complex with globular proteins, we first identified large numbers of complexes between globular proteins and ID segments by using radius-of-gyration-based selection criteria. Consistent with previous studies, we found the interfaces of these complexes to be enriched in hydrophobic residues, and that these residues contribute significantly to the stability of the interaction interface. However, our analyses also show that polar interactions play a larger role in these complexes than in structured protein complexes. Computational alanine scanning and salt-bridge analysis indicate that interfaces in ID complexes are highly complementary with respect to electrostatics, more so than interfaces of globular proteins. Follow-up calculations of the electrostatic contributions to the free energy of binding uncovered significantly stronger Coulombic interactions in complexes harbouring ID segments than in structured protein complexes. However, they are counter-balanced by even higher polar-desolvation penalties. We propose that polar interactions are a key contributing factor to the observed high specificity of ID segment-mediated interactions.
Author Summary
Protein-protein interactions are essential to communication and signal integration in cells. For these processes to be precise, interactions between proteins have to be specific and well coordinated. In order to understand the specificity in protein interactions, researches have focused on interfaces between two or more folded proteins. It has been shown that specificity in interactions between folded proteins relies on shape complementarity, hydrogen bonding, and salt-bridge formation. However, many proteins lack a unique folded structure; the so-called intrinsically disordered proteins. These proteins fluctuate between different conformations in isolation but often adopt a single structure when interacting with partner proteins. As many intrinsically disordered proteins are involved in signaling and regulation, their interactions have to be highly specific. The finding that the interaction interfaces of intrinsically disordered proteins are enriched in hydrophobic residues has led to questions regarding the specificity of interactions mediated by this group of proteins. Here, we show that polar and charged residues play a larger role in interfaces that involve intrinsically disordered proteins compared to interfaces that involve only folded proteins. Our results suggest that polar interactions are key contributors to the specificity of interactions that involve intrinsically disordered proteins.
Related collections
Most cited references52
- Record: found
- Abstract: found
- Article: not found
Gene Ontology: tool for the unification of biology
- Record: found
- Abstract: found
- Article: not found
CHARMM-GUI: a web-based graphical user interface for CHARMM.
- Record: found
- Abstract: found
- Article: not found