- Record: found
- Abstract: found
- Article: found
Synovial DKK1 expression is regulated by local glucocorticoid metabolism in inflammatory arthritis
Read this article at
Abstract
Introduction
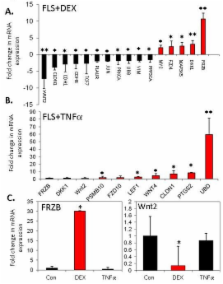
Inflammatory arthritis is associated with increased bone resorption and suppressed bone formation. The Wnt antagonist dickkopf-1 (DKK1) is secreted by synovial fibroblasts in response to inflammation and this protein has been proposed to be a master regulator of bone remodelling in inflammatory arthritis. Local glucocorticoid production is also significantly increased during joint inflammation. Therefore, we investigated how locally derived glucocorticoids and inflammatory cytokines regulate DKK1 synthesis in synovial fibroblasts during inflammatory arthritis.
Methods
We examined expression and regulation of DKK1 in primary cultures of human synovial fibroblasts isolated from patients with inflammatory arthritis. The effect of TNFα, IL-1β and glucocorticoids on DKK1 mRNA and protein expression was examined by real-time PCR and ELISA. The ability of inflammatory cytokine-induced expression of the glucocorticoid-activating enzyme 11beta-hydroxysteroid dehydrogenase type 1 (11β-HSD1) to sensitise fibroblasts to endogenous glucocorticoids was explored. Global expression of Wnt signalling and target genes in response to TNFα and glucocorticoids was assessed using a custom array.
Results
DKK1 expression in human synovial fibroblasts was directly regulated by glucocorticoids but not proinflammatory cytokines. Glucocorticoids, but not TNFα, regulated expression of multiple Wnt agonists and antagonists in favour of inhibition of Wnt signalling. However, TNFα and IL-1β indirectly stimulated DKK1 production through increased expression of 11β-HSD1.
Conclusions
These results demonstrate that in rheumatoid arthritis synovial fibroblasts, DKK1 expression is directly regulated by glucocorticoids rather than TNFα. Consequently, the links between synovial inflammation, altered Wnt signalling and bone remodelling are not direct but are dependent on local activation of endogenous glucocorticoids.
Related collections
Most cited references17
- Record: found
- Abstract: found
- Article: not found
Glucocorticoids act directly on osteoblasts and osteocytes to induce their apoptosis and reduce bone formation and strength.
- Record: found
- Abstract: found
- Article: not found
Bone loss in inflammatory disorders.
- Record: found
- Abstract: found
- Article: not found