- Record: found
- Abstract: found
- Article: found
Identification of a Novel Conjugative Plasmid in Mycobacteria That Requires Both Type IV and Type VII Secretion
Read this article at
ABSTRACT
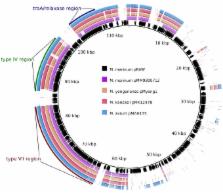
Conjugative plasmids have been identified in a wide variety of different bacteria, ranging from proteobacteria to firmicutes, and conjugation is one of the most efficient routes for horizontal gene transfer. The most widespread mechanism of plasmid conjugation relies on different variants of the type IV secretion pathway. Here, we describe the identification of a novel type of conjugative plasmid that seems to be unique for mycobacteria. Interestingly, while this plasmid is efficiently exchanged between different species of slow-growing mycobacteria, including Mycobacterium tuberculosis, it could not be transferred to any of the fast-growing mycobacteria tested. Genetic analysis of the conjugative plasmid showed the presence of a locus containing homologues of three type IV secretion system components and a relaxase. In addition, a new type VII secretion locus was present. Using transposon insertion mutagenesis, we show that in fact both these secretion systems are essential for conjugation, indicating that this plasmid represents a new class of conjugative plasmids requiring two secretion machineries. This plasmid could form a useful new tool to exchange or introduce DNA in slow-growing mycobacteria.
IMPORTANCE
Conjugative plasmids play an important role in horizontal gene transfer between different bacteria and, as such, in their adaptation and evolution. This effect is most obvious in the spread of antibiotic resistance genes. Thus far, conjugation of natural plasmids has been described only rarely for mycobacterial species. In fact, it is generally accepted that M. tuberculosis does not show any recent sign of horizontal gene transfer. In this study, we describe the identification of a new widespread conjugative plasmid that can also be efficiently transferred to M. tuberculosis. This plasmid therefore poses both a threat and an opportunity. The threat is that, through the acquisition of antibiotic resistance markers, this plasmid could start a rapid spread of antibiotic resistance genes between pathogenic mycobacteria. The opportunity is that we could use this plasmid to generate new tools for the efficient introduction of foreign DNA in slow-growing mycobacteria.
Related collections
Most cited references31
- Record: found
- Abstract: found
- Article: not found