- Record: found
- Abstract: found
- Article: found
GPCRs from fusarium graminearum detection, modeling and virtual screening - the search for new routes to control head blight disease
Read this article at
Abstract
Backgound
Fusarium graminearum (FG) is one of the major cereal infecting pathogens causing high economic losses worldwide and resulting in adverse effects on human and animal health. Therefore, the development of new fungicides against FG is an important issue to reduce cereal infection and economic impact. In the strategy for developing new fungicides, a critical step is the identification of new targets against which innovative chemicals weapons can be designed. As several G-protein coupled receptors (GPCRs) are implicated in signaling pathways critical for the fungi development and survival, such proteins could be valuable efficient targets to reduce Fusarium growth and therefore to prevent food contamination.
Results
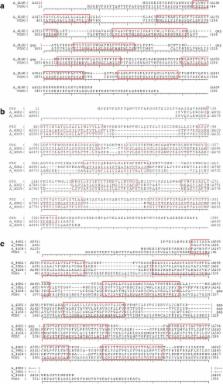
In this study, GPCRs were predicted in the FG proteome using a manually curated pipeline dedicated to the identification of GPCRs. Based on several successive filters, the most appropriate GPCR candidate target for developing new fungicides was selected. Searching for new compounds blocking this particular target requires the knowledge of its 3D-structure. As no experimental X-Ray structure of the selected protein was available, a 3D model was built by homology modeling. The model quality and stability was checked by 100 ns of molecular dynamics simulations. Two stable conformations representative of the conformational families of the protein were extracted from the 100 ns simulation and were used for an ensemble docking campaign. The model quality and stability was checked by 100 ns of molecular dynamics simulations previously to the virtual screening step. The virtual screening step comprised the exploration of a chemical library with 11,000 compounds that were docked to the GPCR model. Among these compounds, we selected the ten top-ranked nontoxic molecules proposed to be experimentally tested to validate the in silico simulation.
Related collections
Most cited references56
- Record: found
- Abstract: found
- Article: not found
Improved protein-ligand docking using GOLD.
- Record: found
- Abstract: found
- Article: not found
Protein structure modeling with MODELLER.

- Record: found
- Abstract: found
- Article: found