- Record: found
- Abstract: found
- Article: found
The genomic substrate for adaptive radiation in African cichlid fish
Read this article at
Abstract
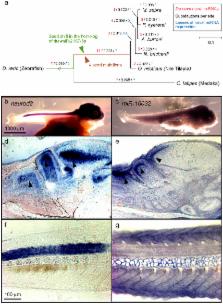
Cichlid fishes are famous for large, diverse and replicated adaptive radiations in the Great Lakes of East Africa. To understand the molecular mechanisms underlying cichlid phenotypic diversity, we sequenced the genomes and transcriptomes of five lineages of African cichlids: the Nile tilapia ( Oreochromis niloticus), an ancestral lineage with low diversity; and four members of the East African lineage: Neolamprologus brichardi/pulcher (older radiation, Lake Tanganyika), Metriaclima zebra (recent radiation, Lake Malawi), Pundamilia nyererei (very recent radiation, Lake Victoria), and Astatotilapia burtoni (riverine species around Lake Tanganyika). We found an excess of gene duplications in the East African lineage compared to tilapia and other teleosts, an abundance of non-coding element divergence, accelerated coding sequence evolution, expression divergence associated with transposable element insertions, and regulation by novel microRNAs. In addition, we analysed sequence data from sixty individuals representing six closely related species from Lake Victoria, and show genome-wide diversifying selection on coding and regulatory variants, some of which were recruited from ancient polymorphisms. We conclude that a number of molecular mechanisms shaped East African cichlid genomes, and that amassing of standing variation during periods of relaxed purifying selection may have been important in facilitating subsequent evolutionary diversification.
Related collections
Most cited references36
- Record: found
- Abstract: found
- Article: not found
The genomic basis of adaptive evolution in threespine sticklebacks
- Record: found
- Abstract: found
- Article: not found
Insights into hominid evolution from the gorilla genome sequence
- Record: found
- Abstract: found
- Article: not found