- Record: found
- Abstract: found
- Article: found
Activation of mitochondrial TUFM ameliorates metabolic dysregulation through coordinating autophagy induction
Read this article at
Abstract
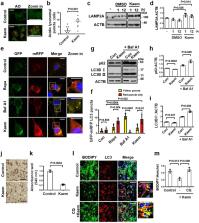
Disorders of autophagy, a key regulator of cellular homeostasis, cause a number of human diseases. Due to the role of autophagy in metabolic dysregulation, there is a need to identify autophagy regulators as therapeutic targets. To address this need, we conducted an autophagy phenotype-based screen and identified the natural compound kaempferide (Kaem) as an autophagy enhancer. Kaem promoted autophagy through translocation of transcription factor EB (TFEB) without MTOR perturbation, suggesting it is safe for administration. Moreover, Kaem accelerated lipid droplet degradation in a lysosomal activity-dependent manner in vitro and ameliorated metabolic dysregulation in a diet-induced obesity mouse model. To elucidate the mechanism underlying Kaem’s biological activity, the target protein was identified via combined drug affinity responsive target stability and LC–MS/MS analyses. Kaem directly interacted with the mitochondrial elongation factor TUFM, and TUFM absence reversed Kaem-induced autophagy and lipid degradation. Kaem also induced mitochondrial reactive oxygen species (mtROS) to sequentially promote lysosomal Ca 2+ efflux, TFEB translocation and autophagy induction, suggesting a role of TUFM in mtROS regulation. Collectively, these results demonstrate that Kaem is a potential therapeutic candidate/chemical tool for treating metabolic dysregulation and reveal a role for TUFM in autophagy for metabolic regulation with lipid overload.
Abstract
Kim, Hwang et al. use in vitro and in vivo models of autophagy disorder/metabolic dysfunction to show that in this context, the natural compound kaempferide is an autophagy enhancer and reveal that one of the underlying mechanisms governing this is mediated by the mitochondrial elongation factor TUFM. This insight may have therapeutic value in the treatment of metabolic disorders.
Related collections
Most cited references63
- Record: found
- Abstract: found
- Article: not found
The coming of age of chaperone-mediated autophagy

- Record: found
- Abstract: found
- Article: found
Mitochondrial electron transport chain, ROS generation and uncoupling (Review)
- Record: found
- Abstract: found
- Article: not found