- Record: found
- Abstract: found
- Article: found
The gene normalization task in BioCreative III
Read this article at
Abstract
Background
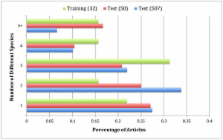
We report the Gene Normalization (GN) challenge in BioCreative III where participating teams were asked to return a ranked list of identifiers of the genes detected in full-text articles. For training, 32 fully and 500 partially annotated articles were prepared. A total of 507 articles were selected as the test set. Due to the high annotation cost, it was not feasible to obtain gold-standard human annotations for all test articles. Instead, we developed an Expectation Maximization (EM) algorithm approach for choosing a small number of test articles for manual annotation that were most capable of differentiating team performance. Moreover, the same algorithm was subsequently used for inferring ground truth based solely on team submissions. We report team performance on both gold standard and inferred ground truth using a newly proposed metric called Threshold Average Precision (TAP- k).
Results
We received a total of 37 runs from 14 different teams for the task. When evaluated using the gold-standard annotations of the 50 articles, the highest TAP- k scores were 0.3297 ( k=5), 0.3538 ( k=10), and 0.3535 ( k=20), respectively. Higher TAP- k scores of 0.4916 ( k=5, 10, 20) were observed when evaluated using the inferred ground truth over the full test set. When combining team results using machine learning, the best composite system achieved TAP- k scores of 0.3707 ( k=5), 0.4311 ( k=10), and 0.4477 ( k=20) on the gold standard, representing improvements of 12.4%, 21.8%, and 26.6% over the best team results, respectively.
Conclusions
By using full text and being species non-specific, the GN task in BioCreative III has moved closer to a real literature curation task than similar tasks in the past and presents additional challenges for the text mining community, as revealed in the overall team results. By evaluating teams using the gold standard, we show that the EM algorithm allows team submissions to be differentiated while keeping the manual annotation effort feasible. Using the inferred ground truth we show measures of comparative performance between teams. Finally, by comparing team rankings on gold standard vs. inferred ground truth, we further demonstrate that the inferred ground truth is as effective as the gold standard for detecting good team performance.
Related collections
Most cited references20
- Record: found
- Abstract: found
- Article: not found
Gene Ontology: tool for the unification of biology
- Record: found
- Abstract: found
- Article: not found
ABNER: an open source tool for automatically tagging genes, proteins and other entity names in text.
