- Record: found
- Abstract: found
- Article: found
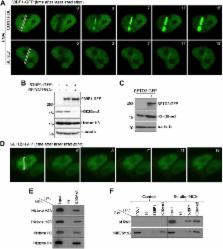
SETD2 is required for DNA double-strand break repair and activation of the p53-mediated checkpoint
Read this article at
Abstract
Histone modifications establish the chromatin states that coordinate the DNA damage response. In this study, we show that SETD2, the enzyme that trimethylates histone H3 lysine 36 (H3K36me3), is required for ATM activation upon DNA double-strand breaks (DSBs). Moreover, we find that SETD2 is necessary for homologous recombination repair of DSBs by promoting the formation of RAD51 presynaptic filaments. In agreement, SETD2-mutant clear cell renal cell carcinoma (ccRCC) cells displayed impaired DNA damage signaling. However, despite the persistence of DNA lesions, SETD2-deficient cells failed to activate p53, a master guardian of the genome rarely mutated in ccRCC and showed decreased cell survival after DNA damage. We propose that this novel SETD2-dependent role provides a chromatin bookmarking instrument that facilitates signaling and repair of DSBs. In ccRCC, loss of SETD2 may afford an alternative mechanism for the inactivation of the p53-mediated checkpoint without the need for additional genetic mutations in TP53.
eLife digest
Normal wear and tear, exposure to chemicals, and ultraviolet light can all damage DNA, so cells rely on a range of sensors and mechanisms to detect and repair damaged DNA. Cells also package DNA molecules inside structures called histones to protect them against damage.
Double-strand breaks—one of the most serious forms of DNA damage—are detected by an enzyme called ATM, and can be repaired in two ways. Bringing the broken strands back together is an obvious method, but it is also error prone. Using templates to generate new DNA to repair the damage is less prone to error, but it can only happen at certain times of the cell cycle.
Some cancers are linked to the faulty repair of double-strand breaks. Moreover, a type of kidney cancer called clear cell renal carcinoma is linked to a lack of activity by a protein called p53, even in individuals who don't have mutations in the gene for this protein. However, many people with this type of cancer have mutations in the gene for a protein called SETD2.
To investigate the links between SETD2 and DNA repair, Carvalho et al. compared cells with and without mutations in the gene for SETD2. It emerged that SETD2 must be present for DNA repair to take place: the SETD2 modifies the histones so that they can recruit the enzymes that repair the DNA via the template approach (which is relatively error free). SETD2 may be particularly important for repairing damage to genes without introducing errors.
Carvalho et al. also show that mutations in SETD2 are sufficient to inactivate p53. The gene for this protein, which impedes the proliferation of cells with genomic aberrations, such as double-strand breaks, is mutated in most cancers. Overall the results help to illustrate how histone modifications and the DNA damage repair mechanisms and checkpoints work in concert to suppress cancer.
Related collections
Most cited references29
- Record: found
- Abstract: found
- Article: not found
The DNA damage response: ten years after.
- Record: found
- Abstract: found
- Article: not found
Human CtIP promotes DNA end resection.
- Record: found
- Abstract: found
- Article: not found