- Record: found
- Abstract: found
- Article: not found
Inhibition of fatty acid oxidation as a therapy for MYC-overexpressing triple-negative breast cancer
Read this article at

Abstract
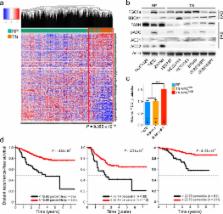
Expression of the oncogenic transcription factor MYC is disproportionately elevated in triple-negative breast cancer (TNBC) compared to estrogen, progesterone and human epidermal growth factor 2 receptor-positive (RP) breast tumors 1, 2 . We and others have shown that MYC alters metabolism during tumorigenesis 3, 4 . However, the role of MYC in TNBC metabolism remains largely unexplored. We hypothesized that MYC-dependent metabolic dysregulation is essential for MYC-overexpressing (MO) TNBC and may thus identify novel therapeutic targets for this clinically challenging subset of breast cancer. Using a targeted metabolomics approach, we identified fatty acid oxidation (FAO) intermediates as being dramatically upregulated in a MYC-driven model of TNBC. A lipid metabolism gene signature was identified in patients with TNBC from The Cancer Genome Atlas (TCGA) database and multiple other clinical datasets, implicating FAO as a dysregulated pathway critical for TNBC metabolism. We find that MO-TNBC displays increased bioenergetic reliance upon fatty acid oxidation (FAO), and that pharmacologic inhibition of FAO catastrophically decreases energy metabolism of MO-TNBC, blocks growth of a MYC-driven transgenic TNBC model and that of MO-TNBC patient-derived xenografts. Our results demonstrate that inhibition of FAO is a novel therapeutic strategy against MO-TNBC.
Related collections
Most cited references31

- Record: found
- Abstract: found
- Article: found
WGCNA: an R package for weighted correlation network analysis

- Record: found
- Abstract: found
- Article: not found
Comprehensive molecular portraits of human breast tumors
- Record: found
- Abstract: found
- Article: not found