- Record: found
- Abstract: found
- Article: found
Identification and characterization of andalusicin: N-terminally dimethylated class III lantibiotic from Bacillus thuringiensis sv. andalousiensis

Read this article at
Summary
Lanthipeptides, ribosomally synthesized and post-translationally modified peptides (RiPPs), can be divided into five classes based on their structures and biosynthetic pathways. Class I and II lanthipeptides have been well characterized, whereas less is known about members of the other three classes. Here, we describe a new family of class III lanthipeptides from Firmicutes. Members of the family are distinguished by the presence of a single carboxy-terminal labionin. We identified and characterized andalusicin, a representative of this family. Andalusicin bears two methyl groups at the α-amino terminus, a post-translational modification that has not previously been identified in class III lanthipeptides. Mature andalusicin A shows bioactivity against various Gram-positive bacteria, an activity that is highly dependent on the α-N dimethylation.
Graphical abstract
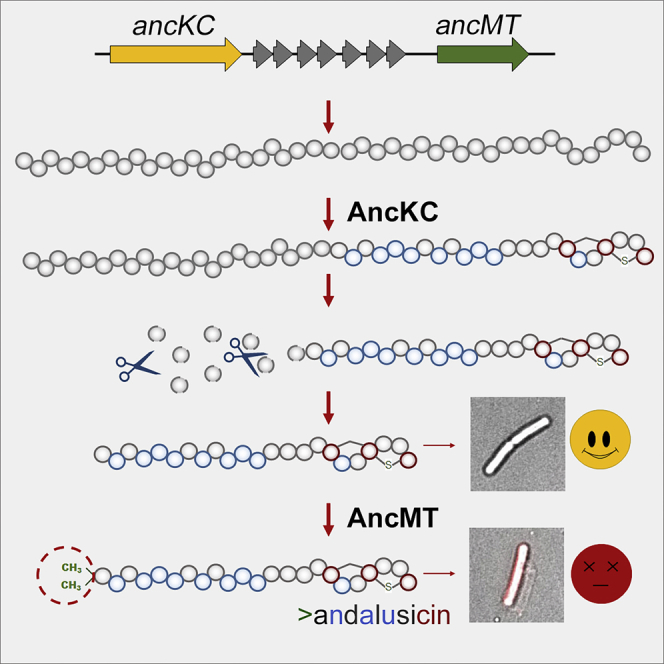
Highlights
Abstract
Chemistry; Biosynthesis; Biochemistry; Medical biochemistry
Related collections
Most cited references55
- Record: found
- Abstract: found
- Article: not found
Cytoscape: a software environment for integrated models of biomolecular interaction networks.
- Record: found
- Abstract: found
- Article: found