
- Record: found
- Abstract: found
- Article: found
Global Biobank analyses provide lessons for developing polygenic risk scores across diverse cohorts

Read this article at
Summary
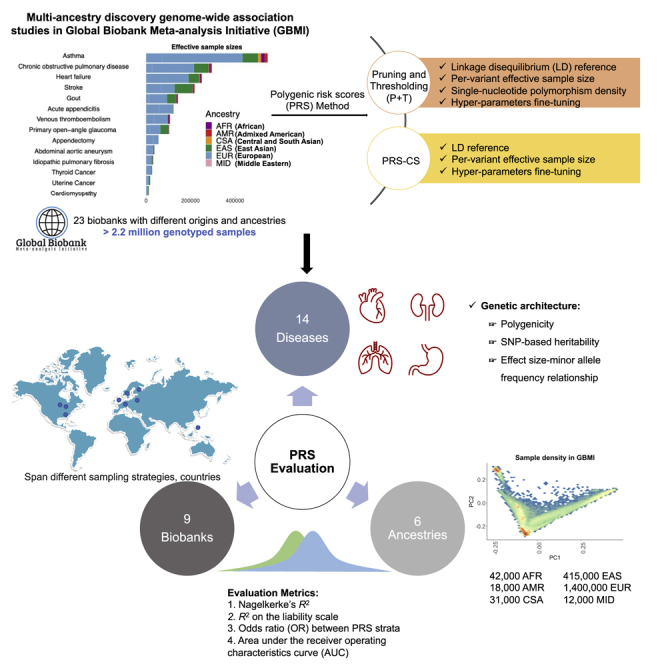
Polygenic risk scores (PRSs) have been widely explored in precision medicine. However, few studies have thoroughly investigated their best practices in global populations across different diseases. We here utilized data from Global Biobank Meta-analysis Initiative (GBMI) to explore methodological considerations and PRS performance in 9 different biobanks for 14 disease endpoints. Specifically, we constructed PRSs using pruning and thresholding (P + T) and PRS-continuous shrinkage (CS). For both methods, using a European-based linkage disequilibrium (LD) reference panel resulted in comparable or higher prediction accuracy compared with several other non-European-based panels. PRS-CS overall outperformed the classic P + T method, especially for endpoints with higher SNP-based heritability. Notably, prediction accuracy is heterogeneous across endpoints, biobanks, and ancestries, especially for asthma, which has known variation in disease prevalence across populations. Overall, we provide lessons for PRS construction, evaluation, and interpretation using GBMI resources and highlight the importance of best practices for PRS in the biobank-scale genomics era.
Graphical abstract
Highlights
Abstract
Wang et al. used the unique resource from Global Biobank Meta-analysis Initiative to develop and evaluate PRSs for 14 disease endpoints with varying genetic architectures and prevalences. They developed guidelines regarding the effects of multi-ancestry and heterogeneous GWASs, trait-specific genetic architecture, and PRS methods on prediction performance across diverse populations.
Related collections
Most cited references58

- Record: found
- Abstract: found
- Article: found
A global reference for human genetic variation

- Record: found
- Abstract: found
- Article: found
Second-generation PLINK: rising to the challenge of larger and richer datasets

- Record: found
- Abstract: found
- Article: found