- Record: found
- Abstract: found
- Article: found
Accuracy of estimated breeding values with genomic information on males, females, or both: an example on broiler chicken
Read this article at
Abstract
Background
As more and more genotypes become available, accuracy of genomic evaluations can potentially increase. However, the impact of genotype data on accuracy depends on the structure of the genotyped cohort. For populations such as dairy cattle, the greatest benefit has come from genotyping sires with high accuracy, whereas the benefit due to adding genotypes from cows was smaller. In broiler chicken breeding programs, males have less progeny than dairy bulls, females have more progeny than dairy cows, and most production traits are recorded for both sexes. Consequently, genotyping both sexes in broiler chickens may be more advantageous than in dairy cattle.
Methods
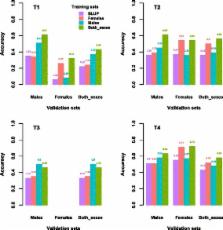
We studied the contribution of genotypes from males and females using a real dataset with genotypes on 15 723 broiler chickens. Genomic evaluations used three training sets that included only males (4648), only females (8100), and both sexes (12 748). Realized accuracies of genomic estimated breeding values (GEBV) were used to evaluate the benefit of including genotypes for different training populations on genomic predictions of young genotyped chickens.
Results
Using genotypes on males, the average increase in accuracy of GEBV over pedigree-based EBV for males and females was 12 and 1 percentage points, respectively. Using female genotypes, this increase was 1 and 18 percentage points, respectively. Using genotypes of both sexes increased accuracies by 19 points for males and 20 points for females. For two traits with similar heritabilities and amounts of information, realized accuracies from cross-validation were lower for the trait that was under strong selection.
Related collections
Most cited references32
- Record: found
- Abstract: found
- Article: not found
Hot topic: a unified approach to utilize phenotypic, full pedigree, and genomic information for genetic evaluation of Holstein final score.
- Record: found
- Abstract: found
- Article: not found