- Record: found
- Abstract: found
- Article: found
Novel Approach to Meta-Analysis of Microarray Datasets Reveals Muscle Remodeling-related Drug Targets and Biomarkers in Duchenne Muscular Dystrophy

Read this article at
Abstract
Elucidation of new biomarkers and potential drug targets from high-throughput profiling data is a challenging task due to a limited number of available biological samples and questionable reproducibility of differential changes in cross-dataset comparisons. In this paper we propose a novel computational approach for drug and biomarkers discovery using comprehensive analysis of multiple expression profiling datasets.
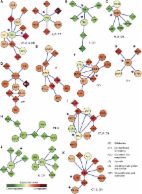
The new method relies on aggregation of individual profiling experiments combined with leave-one-dataset-out validation approach. Aggregated datasets were studied using Sub-Network Enrichment Analysis algorithm (SNEA) to find consistent statistically significant key regulators within the global literature-extracted expression regulation network. These regulators were linked to the consistent differentially expressed genes.
We have applied our approach to several publicly available human muscle gene expression profiling datasets related to Duchenne muscular dystrophy (DMD). In order to detect both enhanced and repressed processes we considered up- and down-regulated genes separately. Applying the proposed approach to the regulators search we discovered the disturbance in the activity of several muscle-related transcription factors (e.g. MYOG and MYOD1), regulators of inflammation, regeneration, and fibrosis. Almost all SNEA-derived regulators of down-regulated genes (e.g. AMPK, TORC2, PPARGC1A) correspond to a single common pathway important for fast-to-slow twitch fiber type transition. We hypothesize that this process can affect the severity of DMD symptoms, making corresponding regulators and downstream genes valuable candidates for being potential drug targets and exploratory biomarkers.
Author Summary
Comparison of gene expression in diseased and normal tissue is a powerful tool of studying processes involved in pathogenesis and searching for potential drug targets and biomarkers of the disease's progression and treatment outcome. We have developed a novel approach for systematic knowledge-driven analysis of gene expression profiling data, which can suggest the underlying cause of the observed differential expression by identifying which expression regulators might be involved. These regulators can not only be the promising subjects of further investigation, but also potential drug targets, as normalization of their activity might alleviate some of the disease's symptoms. The targets downstream of suggested regulators can be proposed as exploratory biomarkers in disease treatment and prognosis. We used our approach to analyze public gene expression datasets of Duchenne muscular dystrophy – a progressive inherited disease in males. Some of the regulators and biomarkers that we found were already investigated in the context of DMD, while some of them were not yet studied and may be of interest for biological and clinical studies.
Related collections
Most cited references75
- Record: found
- Abstract: found
- Article: not found
AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha.
- Record: found
- Abstract: found
- Article: found
Network-based classification of breast cancer metastasis
- Record: found
- Abstract: found
- Article: not found