- Record: found
- Abstract: found
- Article: found
Carotenoid Biosynthesis: Genome-Wide Profiling, Pathway Identification in Rhodotorula glutinis X-20, and High-Level Production

Read this article at
Abstract
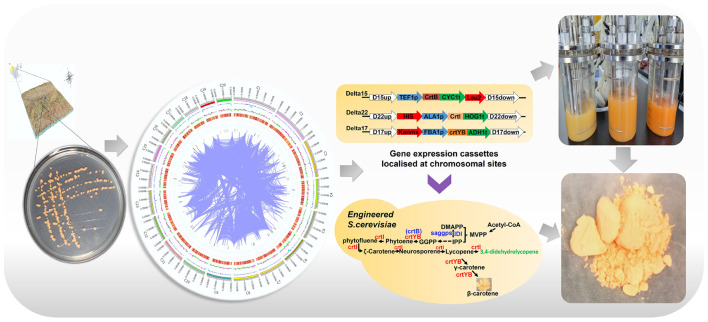
Rhodotorula glutinis, as a member of the family Sporidiobolaceae, is of great value in the field of biotechnology. However, the evolutionary relationship of R. glutinis X-20 with Rhodosporidiobolus, Sporobolomyces, and Rhodotorula are not well understood, and its metabolic pathways such as carotenoid biosynthesis are not well resolved. Here, genome sequencing and comparative genome techniques were employed to improve the understanding of R. glutinis X-20. Phytoene desaturase (crtI) and 15-cis-phytoene synthase/lycopene beta-cyclase (crtYB), key enzymes in carotenoid pathway from R. glutinis X-20 were more efficiently expressed in S. cerevisiae INVSc1 than in S. cerevisiae CEN.PK2-1C. High yielding engineered strains were obtained by using synthetic biology technology constructing carotenoid pathway in S. cerevisiae and optimizing the precursor supply after fed-batch fermentation with palmitic acid supplementation. Genome sequencing analysis and metabolite identification has enhanced the understanding of evolutionary relationships and metabolic pathways in R. glutinis X-20, while heterologous construction of carotenoid pathway has facilitated its industrial application.
Graphical Abstract
Related collections
Most cited references62
- Record: found
- Abstract: found
- Article: not found
MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets.
- Record: found
- Abstract: found
- Article: not found
featureCounts: an efficient general purpose program for assigning sequence reads to genomic features.
- Record: found
- Abstract: found
- Article: not found