- Record: found
- Abstract: found
- Article: not found
Homeostatic MyD88-dependent signals cause lethal inflamMation in the absence of A20
Read this article at

Abstract
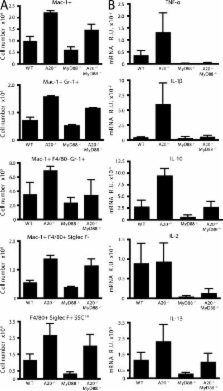
Toll-like receptors (TLRs) on host cells are chronically engaged by microbial ligands during homeostatic conditions. These signals do not cause inflammatory immune responses in unperturbed mice, even though they drive innate and adaptive immune responses when combating microbial infections. A20 is a ubiquitin-modifying enzyme that restricts exogenous TLR-induced signals. We show that MyD88-dependent TLR signals drive the spontaneous T cell and myeloid cell activation, cachexia, and premature lethality seen in A20-deficient mice. We have used broad spectrum antibiotics to demonstrate that these constitutive TLR signals are driven by commensal intestinal flora. A20 restricts TLR signals by restricting ubiquitylation of the E3 ligase tumor necrosis factor receptor–associated factor 6. These results reveal both the severe proinflammatory pathophysiology that can arise from homeostatic TLR signals as well as the critical role of A20 in restricting these signals in vivo. In addition, A20 restricts MyD88-independent TLR signals by inhibiting Toll/interleukin 1 receptor domain–containing adaptor inducing interferon (IFN) β–dependent nuclear factor κB signals but not IFN response factor 3 signaling. These findings provide novel insights into how physiological TLR signals are regulated.
Related collections
Most cited references30
- Record: found
- Abstract: found
- Article: not found
Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis.
- Record: found
- Abstract: found
- Article: not found
Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-18-mediated function.
- Record: found
- Abstract: found
- Article: not found