- Record: found
- Abstract: found
- Article: found
Reduced TRMU expression increases the sensitivity of hair-cell-like HEI-OC-1 cells to neomycin damage in vitro
Read this article at
Abstract
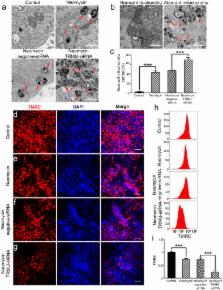
Aminoglycosides are ototoxic to the cochlear hair cells, and mitochondrial dysfunction is one of the major mechanisms behind ototoxic drug-induced hair cell death. TRMU (tRNA 5-methylaminomethyl-2-thiouridylate methyltransferase) is a mitochondrial protein that participates in mitochondrial tRNA modifications, but the role of TRMU in aminoglycoside-induced ototoxicity remains to be elucidated. In this study, we took advantage of the HEI-OC-1 cell line to investigate the role of TRMU in aminoglycoside-induced cell death. We found that TRMU is expressed in both hair cells and HEI-OC-1 cells, and its expression is significantly decreased after 24 h neomycin treatment. We then downregulated TRMU expression with siRNA and found that cell death and apoptosis were significantly increased after neomycin injury. Furthermore, when we down-regulated TRMU expression, we observed significantly increased mitochondrial dysfunction and increased levels of reactive oxygen species (ROS) after neomycin injury, suggesting that TRMU regulates mitochondrial function and ROS levels. Lastly, the antioxidant N-acetylcysteine rescued the mitochondrial dysfunction and cell apoptosis that was induced by TRMU downregulation, suggesting that ROS accumulation contributed to the increased aminoglycosides sensitivity of HEI-OC-1 cells after TRMU downregulation. This study provides evidence that TRMU might be a new therapeutic target for the prevention of aminoglycoside-induced hair cell death.
Related collections
Most cited references46
- Record: found
- Abstract: found
- Article: not found
Mitochondrial transcription factor A regulates mtDNA copy number in mammals.
- Record: found
- Abstract: found
- Article: not found
Essential role of the mitochondrial apoptosis-inducing factor in programmed cell death.
- Record: found
- Abstract: found
- Article: not found